DOI: 10.31038/CST.2019412
Abstract
The association of methylation enzymes with telomerase constitutes a unique abnormality of cancer cells. This abnormality locks methylation enzymes in an exceptionally stable and active state so that hypomethylation of nucleic acids necessary for the cells to undergo Terminal Differentiation (TD) cannot take place. Human body produces metabolites that are able to eliminate telomerase from abnormal methylation enzymes of cancer cells to allow TD to proceed. Cell Differentiation Agent-2 (CDA-2) is a preparation of human metabolites from freshly collected urine, which has been approved for cancer therapy by the Chinese FDA. The effective components of CDA-2 are Differentiation Inducers (DIs) to target on the telomerase of abnormal methylation enzymes and Differentiation Helper Inducers (DHIs) which are the inhibitors of individual enzymes of ternary methylation enzymes. CDA-2 was very effective for the therapy of Myelodysplastic Syndrome (MDS), which is a disease attributable to Cancer Stem Cells (CSCs). We have previously carried out extensive studies on the DHIs of CDA-2.We are now focusing on the DIs of CDA-2 in order to formulate synthetic CDA for the prevention and therapy of cancer via targeting of CSCs.
DIs were purified from CDA-2 solution by procedures including differential solvent extraction, gel filtration, ion exchange chromatography, TLC, and HPLC. The mass of purified active preparation was determined by mass spectroscopy. DI activity was based on the Nitro Blue Tetrazolium (NBT) assay of HL-60 cells.
DIs of CDA-2 were found predominantly as acidic liposomal complexes extractable by dichloromethane. A good proportion of which became covalently linked to inactive carriers which were not soluble in dichloromethane, but soluble in alcohols. We have identified pregnenolone as a DHI of active liposomal complexes. After dissociation from pregnenolone, the active DIs of CDA-2 were not associated with UV absorption peaks of HPLC. We suspected that the active DIs might be acidic peptides derived from endogenous proteins, because we have previously found that acidic peptides of CDA-2 were active DIs. We, thus, randomly picked pentapeptides containing at least two acidic amino acid residues from the sequences of a- and b-hemoglobin for synthesis to test their DI activities. Indeed, acidic pentapeptides of hemoglobin were active as DIs, although the activities were not impressive. Retinoic Acid (RA) and 12-O-TetradecanoylPhorbol-13-Acetate (TPA) are well known DIs with much better activities.
In this study, we found Pyrvinium Pamoate (PP) as the best DHI, and triinosinate + tetrainosinate (I3 + I4) as an acceptable DHI.
With effective DIs and DHIs on hand, our deliberated CDA formulations were as followings: for the therapy of MDS, the CDA-MDS formulation was RA(ED25)-5P-1(ED25)-I3 + I4(RI0.5)-PP(RI0.5)-sodium pregnenolone sulfate(RI0.5); for the therapy of CSCs, the CDA-CSC formulation was RA(ED25)-TPA(ED25)-PP(RI0.5)-resveratrol(RI0.5)-curcumin(RI0.5); for the therapy of brain tumor, the CDA-BT formulation was TPA(2xED25)-PP(2xRI0.5)-sodium phenylbutyrate(RI0.5)-pyrogallol(RI0.5); and for the therapy of melanoma and pancreatic cancer, the CDA-M&P formulation was RA(ED25)-TPA(2xED25)-5P-1(ED25)-PP(2xRI0.5)-sodium tannate(RI0.5). The above CDA formulations all produced 100% NBT + on HL-60 cells.
Keywords
Cancer Prevention and Therapy, Abnormal Methylation Enzymes, Cancer Stem Cells, Synthetic CDA Formulations, Differentiation Inducers, Differentiation Helper Inducers.
Introduction
Methylation enzymes play a critical role on the regulation of cell replication and differentiation, because DNA methylation controls the expression of tissue specific genes 1] and pre-rRNA ribose methylation controls the production of ribosomes 2], which in turn dictate the commitment of cells to initiate replication 3]. If enhanced production of ribosomes is locked in place, it becomes a factor to drive carcinogenesis [4]. Biological methylation is mediated by a ternary enzyme complex consisting of Methionine AdenosylTransferase (MAT)-MethylTransferase (MT)-S-Adenosyl Homocysteine Hydrolase (SAHH) [5,6]. These enzymes must be in the ternary enzyme complex to become stable and functional. In the monomeric state, individual enzymes are quickly inactivated. SAHH is the most unstable enzyme, followed by MT, and then MAT. MTs in the monomeric state have a great tendency to be converted into nucleases to trigger apoptosis. SAHH requires a steroid factor to assume a configuration favorable for the formation of dimeric enzyme complex with MT, which is then in a position to form ternary enzyme complex with MAT. In steroid hormone target tissues such as prostate and breast, steroid hormones are the stabilizing factors of SAHH. Other tissues require similar steroid factors generated by the growth signals to stabilize SAHH [7]. In normal cells, steroid factors are the dominant factors to regulate methylation enzymes. In cancer cells and telomerase expressing primitive stem cells such as embryonic stem cells and progenitor stem cells, MAT is associated with telomerase [8], which becomes the dominant factor to regulate methylation enzymes. The association of MATL, the low Km normal isozyme of MAT, with telomerase changes the kinetic properties of MATL and the regulation of methylation enzymes. Km values of MATL and MATLT, the telomerase associated tumor isozyme, are 3 µM and 20 µM methionine, respectively, and those of SAHHL and SAHHLT are 0.3 µM and 2 µM adenosine, respectively [5, 6, 8]. The increased Km value of MATLT suggests that methylation enzymes of cancer cells have elevated levels of bound S-adenosylmethionine (AdoMet). According to Prudova et al. [9], the binding of AdoMet to a protein could protect that protein against protease digestion. It appears then that the increased pool size of AdoMet in cancer cells is very important for the stability, and therefore the activity of methylation enzymes to promote malignant growth. Chiba et al. [10] found that the pool sizes of AdoMet and S-adenosylhomocysteine (AdoHcy) shrunk greatly when cancer cells were induced to undergo TD. This finding strongly supports our arguments that the association of telomerase with methylation enzymes greatly increase the stability and the activity of methylation enzymes of cancer cells so that hypomethylation of nucleic acids required for the cell to undergo TD cannot take place [6,11]. Thus, it is very convincing that abnormal methylation enzymes play a critical role on the evolution and the progression of cancer.
Association of telomerase with methylation enzymes locks methylation enzymes in extremely stable and active state to block cell differentiation. Telomerase is actively expressed in embryonic stem cells and progenitor stem cells. Differentiation of telomerase expressing normal stem cells will be blocked like cancer cells. There is another way to achieve DNA demethylation bypassing the differentiation blockade created by abnormal methylation enzymes. Tet dioxygenases carry out oxidation of 5 mC to generate 5 hmC, 5 fC and 5 caC [12–15], and 5 caC is finally replaced with C by thymine DNA glycosylase [16,17]. 5 hmC is the stable intermediate in the oxidative demethylation of 5 mC [18]. So far three Tet dioxygenases have been identified. Tet 1 preferentially acts on the 5 mC located at the transcriptional start site, whereas Tet 2 preferentially acts on the 5 mC located in the gene body [19]. Tet 3 is expressed at very high levels in oocytes and zygotes, with rapidly declining at the two cell stage. Tet 3 is responsible for the erase of paternal 5 mC in fertilized oocytes [20]. Tet enzymes are very active in embryonic stem cells to direct extraembryonic lineage differentiation [21,22]. These enzymes are frequently mutated to become dysfunctional or silenced in cancer cells [23–26]. The expression and function of Tet enzymes marks the difference between normal primitive stem cells and cancer cells. Consequently, destabilization of abnormal methylation enzymes is the only option workable to induce TD of cancer cells.
Destabilization of abnormal methylation enzymes is a very effective strategy for cancer therapy. The therapy of acute promyelocytic leukemia (APL) with RA yielded a stunning complete remission around 90% [27]. The remission, however, was only transient. Most patients relapsed within a year, and became resistant to further treatment [28, 29]. A combination of RA and As2O3 produced a more satisfactory long lasting remission [29]. RA is a DI, and As2O3 is a DHI [7]. DI is the chemical capable of eliminating the association of telomerase from abnormal methylation enzymes, and DHI is the inhibitor of individual enzymes of ternary methylation enzymes. It appears then that a combination of DI and DHI is necessary to make a perfect drug for cancer therapy. DHI alone can be very effective for cancer therapy too. After all, human body is producing DIs, although cancer patients are unable to retain such valuable metabolites in the body. Excess DHI can salvage the loss of endogenous DIs. Imatinib mesylate is the standard care for chronic myeloid leukemia [30], which is another good example of effective cancer therapy by destabilization of abnormal methylation enzymes. Signal transduction inhibitors such as imatinib mesylate are excellent DHIs [7]. Phenylbutyrate was our creation of the first DHI which was only modestly active requiring mM concentrations to function,[31]. Nevertheless, it has demonstrated therapeutic efficacy on often untreatable brain tumors [32,33]. Brain compartment is protected by blood brain barrier. The loss of endogenous DIs is not as severe as other body compartments. Therefore, even modest DHIs can exercise good therapeutic effects. The therapeutic effect of phenylbutyrate was greatly enhanced when it was used in combination with signal transduction inhibitors [34,35], which were much better DHIs effective in µM concentrations [7]. DHIs, such as dietary polyphenols, are frequently suggested for chemoprevention of cancer [36–39].
In 1987, Liau et al. [40] brought up chemosurveillance as a natural defense mechanism against cancer. This hypothesis was based on the observation that healthy people could maintain a steady level of hydrophobic metabolites in their plasma, whereas cancer patients tended to show deficiency of such metabolites due to excessive urinary excretion [41]. Among such metabolites are chemicals capable of inducing cancer cells to undergo TD [42,43]. The evolution of cancer in the case of MDS strongly supports the validity of this hypothesis.
MDS often starts with a display of immunological disorders associated with inflammation [44], which prompts the production of inflammatory cytokines. Among such cytokines, TNF is a critical factor related to the development of MDS [45]. It causes excessive apoptosis of bone marrow stem cells, thus severely affects the ability of the patient to produce hematopoietic cells such as erythrocytes, platelets, and neutrophils. TNF is also named cachectin, because of its involvement in the symptom known as cachexia. Cachexia is a symptom commonly shared by inflammatory patients and cancer patients. A characteristic disorder of cachexia is the excessive urinary excretion of low molecular weight metabolites because of vascular hyperpermeability cause by TNF [46,47]. As a consequence, chemosurveillance normally operating in healthy people to keep a check on progenitor stem cells is disrupted under pathophysiological conditions created by TNF to allow progenitor stem cells to buildup in order to replenish unipotent stem cells wiped out by TNF. The high levels of telomerase in the peripheral and bone marrow leukocytes in MDS patients is an indication of the widespread multiplication of progenitor stem cells which express telomerase [48,49]. During the course of MDS progression, mutations on Tet2, DNMT3A, IDH1/2, ASXL1, EZH2, and RNA splicing enzymes are frequently observed [50–54], which may play a significant role on the evolution of progenitor stem cells to become CSCs [55]. As anemia symptom becomes worse, chromosomal abnormalities such as translocation and deletion characteristic of cancer cells set in to speed up replication eventually pushing MDS patients to become acute myeloid leukemia patients [56–59].
Vidaza and decitabine are the two hypomethylating agents approved for the therapy of MDS in the USA. CDA-2 is a hypomethylating agent approved by China for the therapy of cancer,[60]. Vidaza and decitabine achieve DNA hypomethylation by promoting covalent bond formation between DNA Methyltransferase (DNMT) and the azacytosine base incorporated into DNA to titrate out DNMT [61], whereas CDA-2 achieves DNA hypomethylation by converting abnormal methylation enzymes into normal enzymes [6,43]. An aborted clinical trial of CDA-2 on MDS was conducted on 117 patients in China. Based on two cycles of treatment protocols, CDA-2 yielded a slightly better therapeutic efficacy under cytological evaluation, and a marked better therapeutic efficacy under hematological improvement evaluation in comparison to vidaza and decitabine,[62, 63]. Apparently CDA-2 had a better therapeutic effect and devoid of serious adverse side effects, whereas decitabine was a proven carcinogen [64]. Since MDS is a disease attributable to CSCs [55], synthetic CDA formulations ought to do well on CSCs.
CSCs constitute only a small subpopulation within a tumor. These cells, nevertheless, are now thought to confer many of adverse characteristics that contribute to treatment failure,[65–69]. Many biological characteristics that enable cancer progression are attributable to CSCs, including angiogenesis, metastasis, recurrence, and drug resistance. Elimination of CSCs is, therefore, very critical to the success of cancer therapy. CSCs are both resistant to cytotoxic chemotherapy and radiotherapy, because these cells overexpress ATP binding cassette drug pumps, and are mostly in dormant state unresponsive to radiation [70–73]. CSCs are equivalent to progenitor stem cells of normal organs or tissues, which replicate only in response to developmental or pathological needs, e.g. growth or wound healing. Thus, CSCs are very responsive to induction of differentiation, which may be the most effective approach to target CSCs.
The objective of this study is to use CDA-2 as a model to develop synthetic CDA formulations for the prevention and therapy of cancer via targeting of CSCs. CDA-2 is a preparation of natural hydrophobic metabolites purified from freshly collected urine by reverse phase chromatograph [74]. We have carried out extensive studies on DHIs of CDA-2 [7,31,74–77]. We are now focusing on DIs of CDA-2, which are the most important active components of CDA-2. When DIs becomes available, we will be in a position to solve problems brought up by CSCs.
Methods and Materials
Chemicals and Reagents
Chemicals, chromatographic supplies, and cell culture supplies were purchased from Sigma, St. Louise, MO, unless otherwise indicated. 35×10 mm cell culture dishes were purchase from CytoOne, USA Scientific. Com. Sep-Pak C18 cartridges were purchased from Walters Associates, Milford, MA. Cosmocil C18 column was purchased from Nacalai Tesque, Kyoto, Japan. Acidic pentapeptides were purchased from GenicBio Company of Shanghai, China. 1.5 liter of CDA-2 solution, 304 mg/ml, was a gift of Mr. Zhanji Sun, the general manager of NT Pharmaceuticals, Jiangsu Co. Ltd, China.
Culture of HL-60 Cells
HL-60 cells were purchased from ATCC, Manassas, VI, which were initially maintained in ISCOVE’s modified medium, supplemented with 10% fetal bovine serum, 2 mM glutamine, 50 units/ml penicillin-50 µg/ml streptomycin for a few generations, and then transferred to RPMI 1640 medium to replace ISCOVE’s modified medium. Cells were subcultured every 3 to 4 days at an initial concentration of 5–10 × 104 cells/ml.
NBT assay
NBT assay was conducted as previously described [7]. Each 35×10 mm cell culture dish contained 2 ml of RPMI 1640 culture medium. HL-60 cells at an initial concentration of 5–10 × 104 cells were incubated with or without drugs for 3 days. Approximately 2.5×105 cells were precipitated at 600xg for 5 min. The cell pellet was suspended in 3 drops from a Pasteur pipet of NBT reagent consisting 1 mg NBT and 5 µg TPA per ml Hank balanced salt solution (HBSS), and incubated at 37o C for 30 min. The reaction was terminated by the addition of a drop from a Pasteur pipet of 4% paraformaldehyde in HBSS. NBT + cells were counted under microscope using a hemacytometer.
Determination of potency of DHIs
The potency of DHIs was assessed by the reductive index as previously described [7]. Cell culture dishes were divided into several sets of 5 dishes containing RA of different concentrations to induce between 0 to 60% NBT + . One set had RA alone as control to yield Effective Dosage50 (ED50) of RA. Other sets had different concentrations of DHIs together with RA concentrations matching the control set. After incubation at 37o C for 72 h, cell numbers from each dish were counted, and an aliquot was withdrawn for NBT assay as above described. NBT + cells in the control dishes without any drug were always below 1%. In the presence of different DHIs alone, NBT + cells in general were below 10%. The respective control value was subtracted from each experimental value to yield the actual ED value. ED50 value, defined as the dosages that induced 50% NBT + cells, were estimated from plots of NBT + values versus concentrations of RA in the absence and presence of DHIs. The Reductive Index (RI) is defined as the ED50 in the presence of DHI divided by the ED50 value of RA alone. The value is inversely related the effectiveness of the DHI agent.
Bio-Gel P2 gel filtration
DIs preparation in less than 4 ml was put onto a Bio-Gel P2 column, 2.5×95 cm for gel filtration resolution. The elution was carried out by 25 mM phosphate buffer, pH 7.8, collecting 4.2 ml/tube/5 min. An aliquot from each tube was withdrawn to dilute with 1 ml of H2O for the determination of A280 absorption, and another aliquot was withdrawn from the filtrate of 0.2 µm membrane filter for the determination of NBT + .
TLC Chromatography
DIs of different Kav fractions from Bio-Gel P2 gel filtration were recovered by C18 cartridge. The fraction was acidified to pH 2.5 and passed the solution through a C18 cartridge. The cartridge was washed twice with 5 ml H2O, and then eluted with 3 ml of 80% methanol. The methanol eluant was evaporated to dryness in a rotary evaporator. The residue was dissolved in a small amount of methanol to apply to a plate of silica gel. The chromatography was developed by ascending chromatography with BuOH-HoAc-H2O (9:2:4) for 8 h to allow the solvent to travel for 17.5 cm from the origin. The silica plate was air dried in a hood overnight. UV images were marked by a pencil. The bands were scraped off the plate with a spatula to put into centrifuge tubes for the extraction with methanol. The methanol extract was evaporated to dryness in a rotary evaporator, and the residue was dissolved in a small amount of methanol for the determination of A280 and NBT + .
Sephadex LH20 Chromatography
A methanol solution of DI subfraction in less than 4 ml was put onto a Sephadex LH20 column, 2.5×36 cm, for chromatography. The elution was carried out by methanol, collecting 2.8 ml/tube/2 min. An aliquot from each tube was withdrawn to dilute with 1 ml of methanol for the determination of A280 absorption, and another aliquot was withdrawn for the determination of NBT + .
DEAE-Sephadex Chromatography
An aqueous solution of DI subfraction with NaCl concentration less than 25 mM and pH 7.8 was put onto a DEAE-Sephadex column, 1.4×27 cm, for chromatography. The column was initially washed with H2O until A280 absorption was no longer detectable. The column was then eluted with 160 ml of a linear gradient of NaCl from 0 to 2 M, collecting 4 ml/tube/4 min. An aliquot from each tube was withdrawn to dilute with l ml of H2O for the determination of A280 absorption, and another aliquot was withdrawn from the filtrate of 0.2 µm membrane filter for the determination of NBT + .
HPLC
A 50 µl of active DI preparation was injected into a Cosmosil (5C18-RA-II) column, 4.6×250 mm, for HPLC resolution using Hewlett Packard 1050 instrument. The flow rate was set at 0.5 ml/min. The elution during the initial 10 min was carried out by a linear gradient of solution A (5% HoAc) from 100% to 0%, and solution B (80% methanol) from 0% to 100%. Thereafter, the column was eluted with solution B until no more A280 absorption was detectable. HPLC fractions were evaporated to dryness in a rotary evaporator. The residue was redissolved in a small volume of methanol for the determination of NBT + .
Mass Spectroscopy
HPLC purified DHI from CDA-2 was injected into LC-MS instrument for the determination of mass.
Results
Purification of DIs from CDA-2 solution
Obviously active DIs and DHIs of CDA-2 are making a significant contribution to protect the vast majority of healthy people from becoming cancer patients. We ought to study these active metabolites which are doing a big favor to benefit human being.
When CDA-2 solution, 304mg/ml, pH 6.7, was acidified to pH 2 with 2N HCl, 32% of A280 absorption and almost 100% of DIs were found in the sediment collected by centrifugation at 1200xg for 30 min. The sediment was 50 ml of very dark colored viscous liquid from 1.5 liter of CDA-2 solution. The sediment was first extracted 3 times with 2 volumes of dichloromethane in a flask by shaking overnight each time in a shaker. Dichloromethane extract was the orange solution of the upper phase, which was poured off the flask. Dichloromethane extracted 16.1% of the A280 absorption and 37.6% of the DI activity from the pH 2 sediment of CDA-2 solution. Next, the remaining sediment was dissolved in 5 volumes of methanol, and insoluble materials were removed by centrifugation at 1200xg for 30 min. Methanol extracted 69.7% of the A280 absorption and 60% of the DI activity from the pH 2 sediment of CDA-2 solution. Methanol insoluble residue was dissolved in dilute NaOH solution, and pH adjusted to 7.8, which constituted 14.1% of the A280 absorption and 2.4% of the DI activity of the pH 2 sediment of CDA-2 solution. DIs soluble in dichloromethane are very hydrophobic metabolites and DIs soluble in methanol are less hydrophobic.
Gel filtration profiles of DIs on Bio-Gel P2 column chromatography
Organic solvents of DI extracts were removed by rotary evaporator at temperature below 60o C. The residues were dissolved in dilute NaOH solution and pH adjusted to 7.8. 4 ml aliquot from each preparation containing approximate 100,000 A280 absorption units was put on a column of Bio-Gel P2 column for gel filtration as described in Methods and Materials. Dichloromethane extractable DIs have two major peaks at Kav = 0.43 and 0.52, and a minor peak at Kav = 1.45 as shown in Fig. 1. Kav = 0.43 and 0.52 are quite large molecular weight complexes.
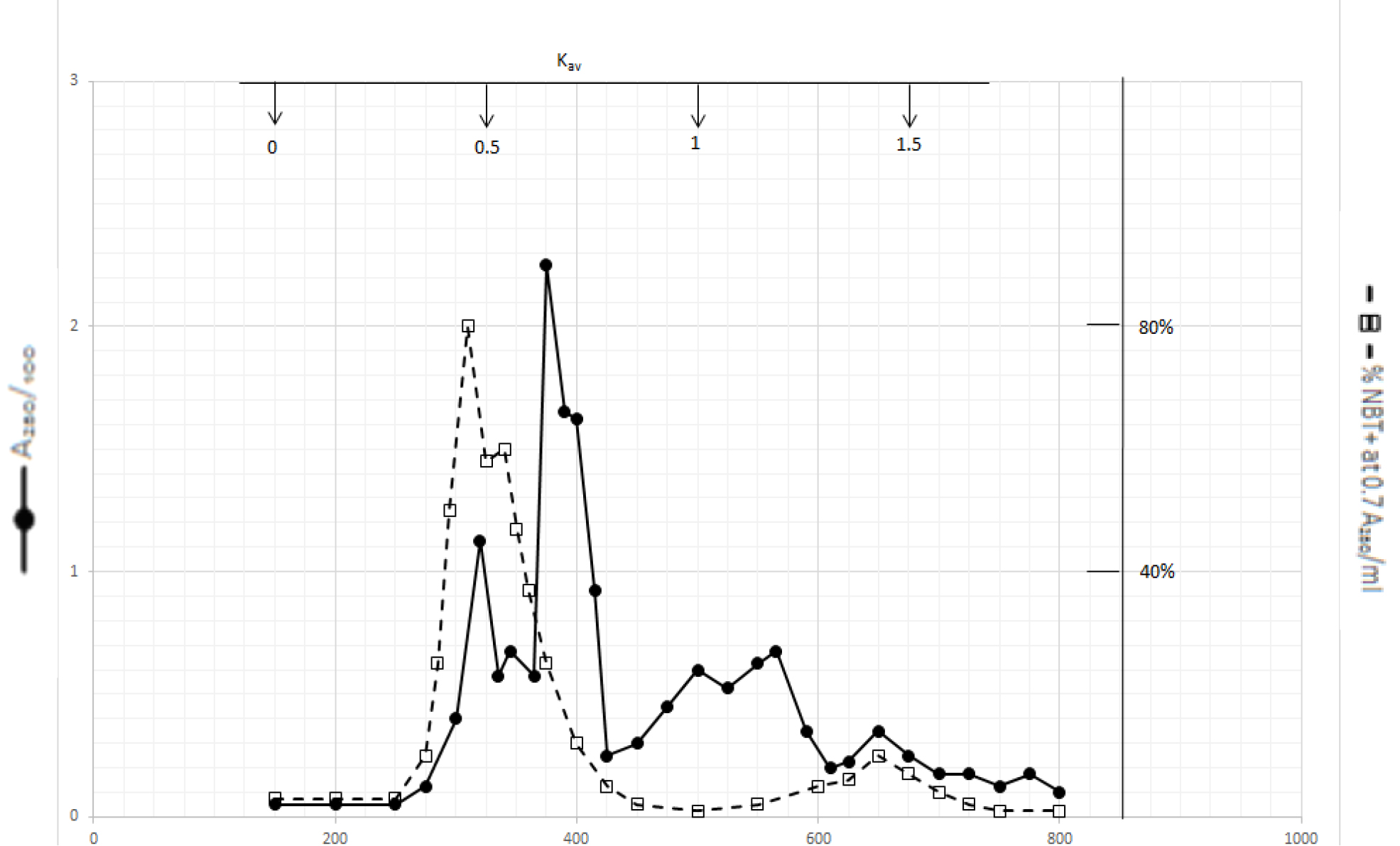
Figure 1. Gel filtration profile of the dichloromethane extractable DIs of CDA-2
4 ml of dichloromethane extractable DIs of CDA-2 containing approximately 100,000 A280 absorption units were put onto a column of Bio-Gel P2 column for gel filtration as described in Methods and Materials.
Gel filtration profile of methanol extractable DIs is presented in Fig. 2. Methanol extractable DIs have a major peak at Kav = 0 and two minor peaks at Kav = 0.43 and 0.52 which are probably the major components of dichloromethane extractable DIs. DIs of Kav = 0 are even bigger than those of dichloromethane extractable DIs
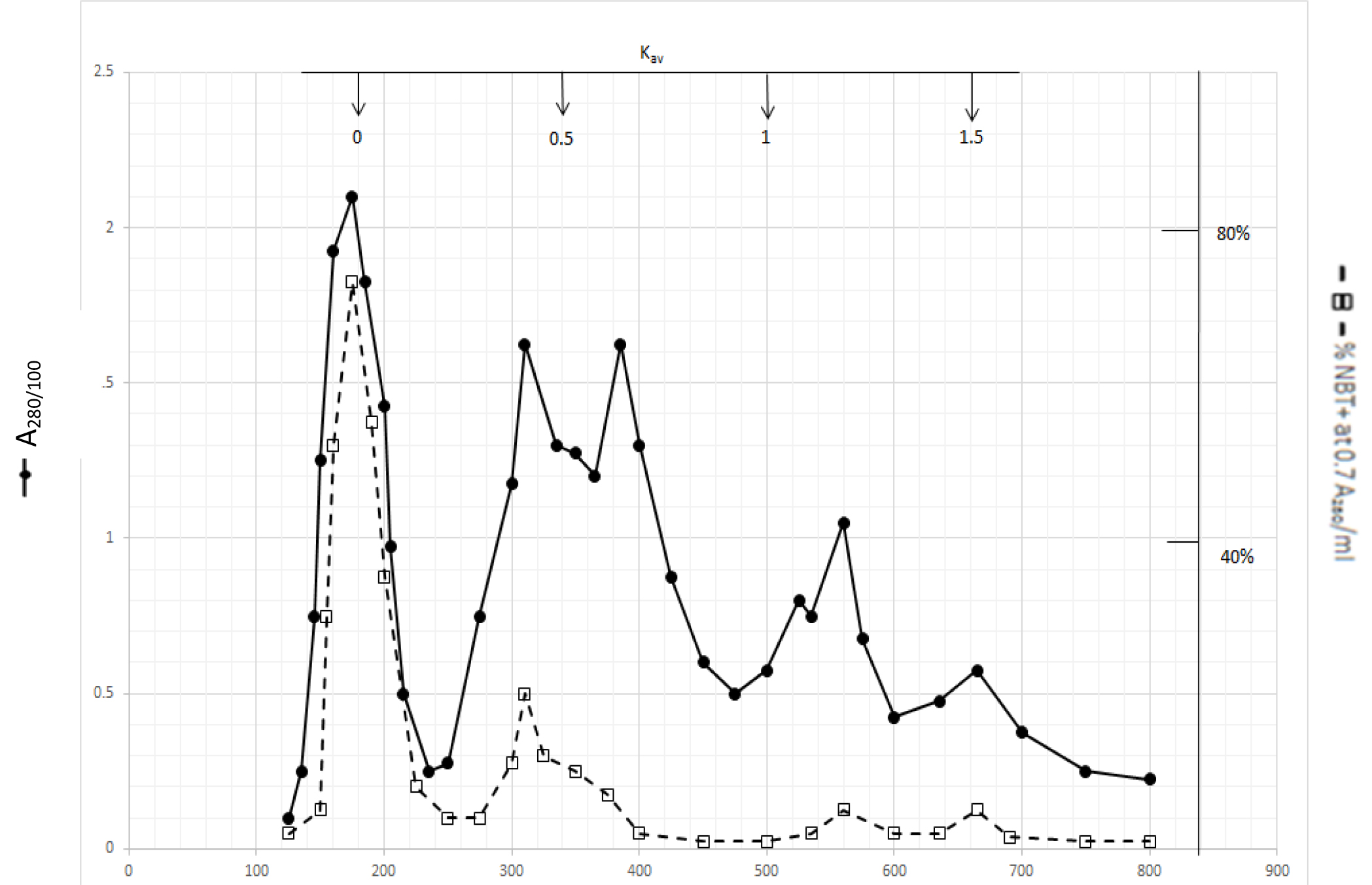
Figure 2. Gel filtration profile of the methanol extractable DIs of CDA-2
4 ml of methanol extractable DIs of CDA-2 containing approximately 100,000 A280 absorption units were put onto a column of Bio-Gel P2 column for gel filtration as described in Methods and Materials.
The dark solution of the Kav = 0 of methanol extractable DIs produced scale-like dark precipitate and clear light color solution when the pH was acidified to 2 with 2N HCl. DI activities became dissociated from the dark inactive carrier when the precipitate was dissolved in 0.5N NaOH and incubated at 37o C as shown in Fig. 3.
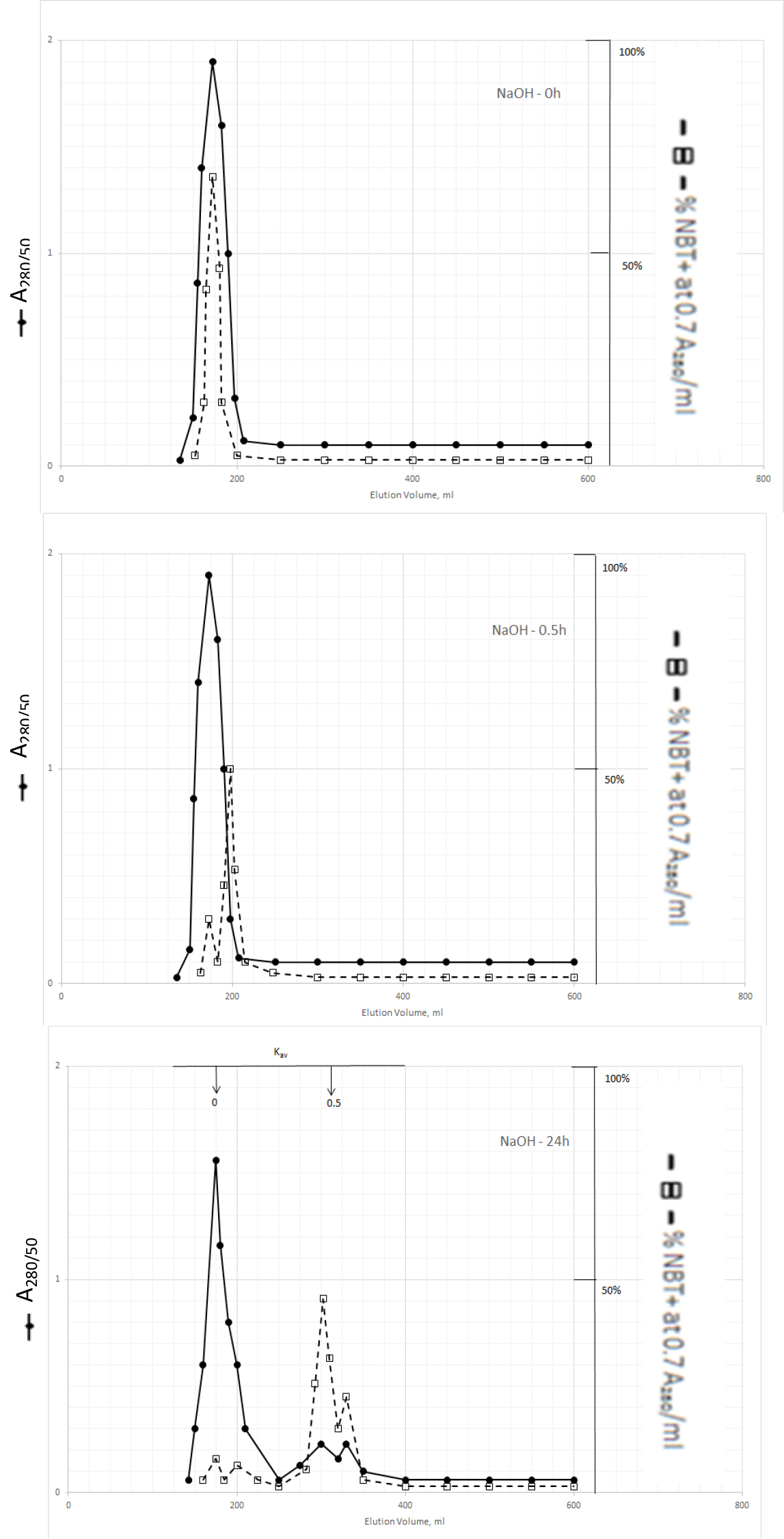
Figure 3. Gel filtration profiles of the DIs of Kav = 0 of Fig. 2 upon incubation with 0.5N NaOH at 37o C
DIs of Kav = 0 were obtained from Fig. 2 by adjusting the active fractions to pH 2 with 2N HCl. The active DIs became scale like dark precipitate, which was collected by centrifugation at 1200xg for 30 min. The residue was dissolved in 6 ml of 0.5N NaOH, 2 ml aliquot containing a total of 1540 A280 absorption units was withdrawn to immediately adjusted to pH 7 for gel filtration as described in Fig. l. Another 2 ml aliquot was withdrawn after incubation at 37o C or 0.5 h to be neutralized for gel filtration. And the last 2 ml was neutralized for gel filtration after incubation at 37o C for 24 h.
The major DI peak shifted from Kav = 0 to Kav = 0,09 after incubation at 37oC for 0.5 h, and then to Kav = 0.43 and 0.52 after incubation for 24 h. The Kav = 0.43 and 0.52 DIs were extractable by dichloromethane when the solution was acidified to pH 2. It appears then that DIs of Kav = 0.43 and 0.52 are the major DIs of CDA-2. A good proportion of these DIs becomes covalently linked to the inactive carrier of Kav = 0 previously identified as pigment peptides,[43].
TLC chromatography of dichloromethane extractable DIs of CDA-2
Dichloromethane extractable DIs of CDA-2 from Fig. 1 were recovered by C18 cartridges and resolved by TLC as described in Methods and Materials. Result is presented in Table 1. Significant DI activities were widespread detectable from Rf = 0.15 to 0.92. Similar widespread activities were also found on DIs of Kav = 0.52. The only difference was the relative activity of different Rf bands. DIs of Kav = 0.43 had more DIs with higher Rf values. DIs of Kav = 1.45 were much simple. Rf = 0.57 band stood out as the only major active band. TLC data serve to indicate that DIs of Kav = 0.43 and 0.52 are complexes of multiple different DIs or a few limited DIs in association with multiple different DHIs or inactive materials.
Table 1. TLC chromatography of the Kav=0.43 DIs of Fig. 1
|
Rf
|
UV images
|
A280/ml
|
% Growth
|
% NBT +
|
|
0.95
|
Bright blue fluorescent
|
0.05
|
73
|
5.8
|
|
0.92
|
Gold fluorescent
|
0.16
|
59
|
22.8
|
|
0.86
|
Dark absorption
|
0.23
|
77
|
7.4
|
|
0.81
|
Gold fluorescent
|
0.21
|
54
|
15.8
|
|
0.74
|
Gold fluorescent
|
0.23
|
68
|
5.3
|
|
0.62
|
Gold fluorescent
|
0.19
|
90
|
11.6
|
|
0.57
|
Dark absorption
|
0.16
|
81
|
44.0
|
|
0.44
|
Bright blue fluorescent
|
0.15
|
47
|
17.5
|
|
0.34
|
Multiple narrow gold fluorescent
|
0.10
|
61
|
17.9
|
|
0.15
|
Weak gold fluorescent
|
0.05
|
73
|
11.5
|
|
0.08
|
Very weak gold fluorescent
|
0.05
|
85
|
4.3
|
Methanol solution of Kav=0.43 DIs of Fig. 1 was applied to a TLC plate for chromatography as described in Methods and Materials.
Sephadex LH20 Chromatography
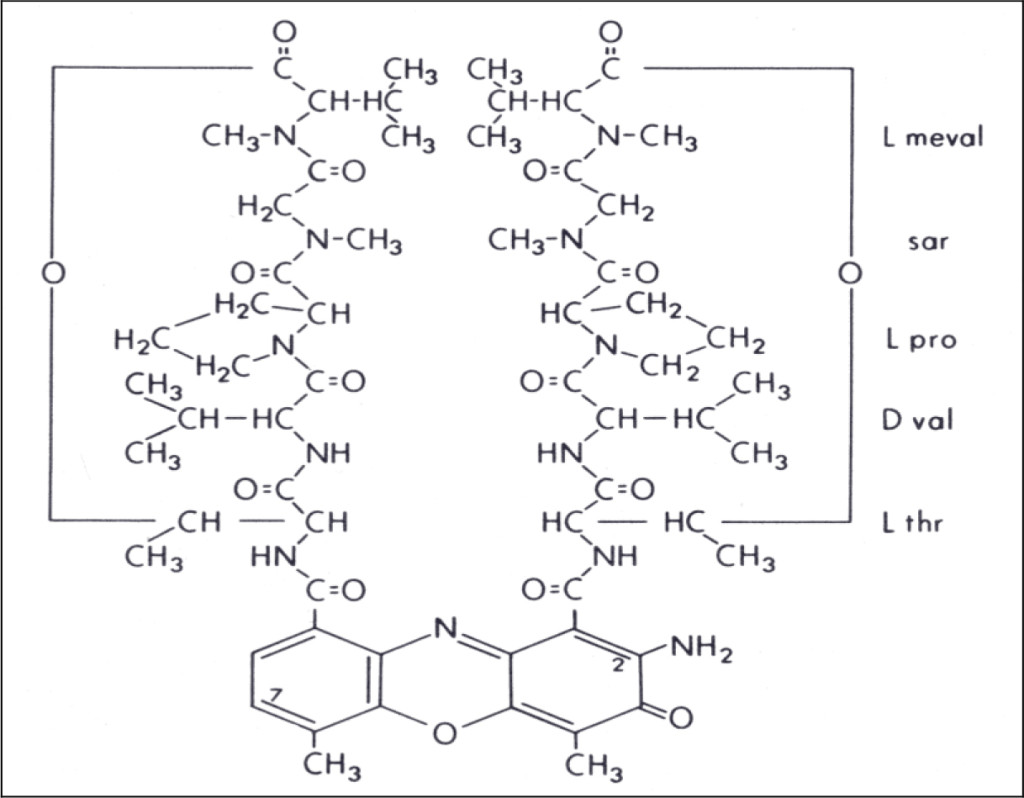
DIs of Kav = o.43 and 0.52 from Fig. 1 were recovered by C18 cartridges as above described. 4 ml of methanol solution containing approximately 25,000 A280 absorption units were put on a column of Sephadex LH20 column for chromatography as described in Methods and Materials. The chromatographic profile is shown in Fig. 4. Two active peaks similar to two Bio-Gel P2 peaks of Kav = 0.43 and 0.52 were found, one eluted between 150–180 ml, and the other between 180–210 ml. The separation of two peaks on Sephadex LH 20 chromatography appeared more clean cut than those on Bio-Gel P2 gel filtration. The active fractions of two peaks were pooled separately, and methanol was removed by rotary evaporator. The residue of each active fractions was suspended in 5 ml H2O, and 1N NaOH was added dropwise to bring pH up to 7.8 to dissolve the residue. NaOH soluble material gave rise to a total of 11, 500 A280 absorption units from 150–180 ml peak, and 5,500 A280 absorption units from 180–210 ml peak. These preparations were separately put on a column of DEAE-Sephadex for chromatography as described in Methods and Materials. DEAE-Sephadex chromatographic profiles are shown in Fig. 5 and 6. The 150–180 ml peak of Sephadex LH20 chromatography gave rise to two active fractions eluted between 0.55–0.75 M NaCl as a major peak and between 0.85–1.05 M NaCl as a minor shoulder peak. The 180–210 ml peak of Sephadex LH 20 chromatography produced a reverse profile: a minor shoulder peak between 0.55–0.75 M NaCl and a major peak between 0.85–1.05 M NaCl. When DEAE-Sephadex chromatographic fractions were stored at 4o C for more than 1 week, noticeable precipitate was found in active fractions from 0.55–1.05 M NaCl. The precipitate was inactive as DIs. The DI activity of the supernatant declined greatly to 22% of the original activity, which was restored to almost the original activity when enough precipitate redissolved in methanol was added back. Thus, the precipitate fits the description as DHI [31]. Upon purification by HPLC, the precipitate yielded a major UV peak at RT-19,1. The eluant of the RT-19.1 peak was evaporated to dryness by rotary evaporator, and redissolved in a small volume of methanol. Subsequent HPLC analysis showed a single RT-19.1 peak as shown in Fig. 7. The mass determination of the RT-19.l HPLC peak of Fig. 7 yielded a mass of 316.118 as shown in Fig. 8. This mass is very close to the mass of pregnenolone, which is 316.48.
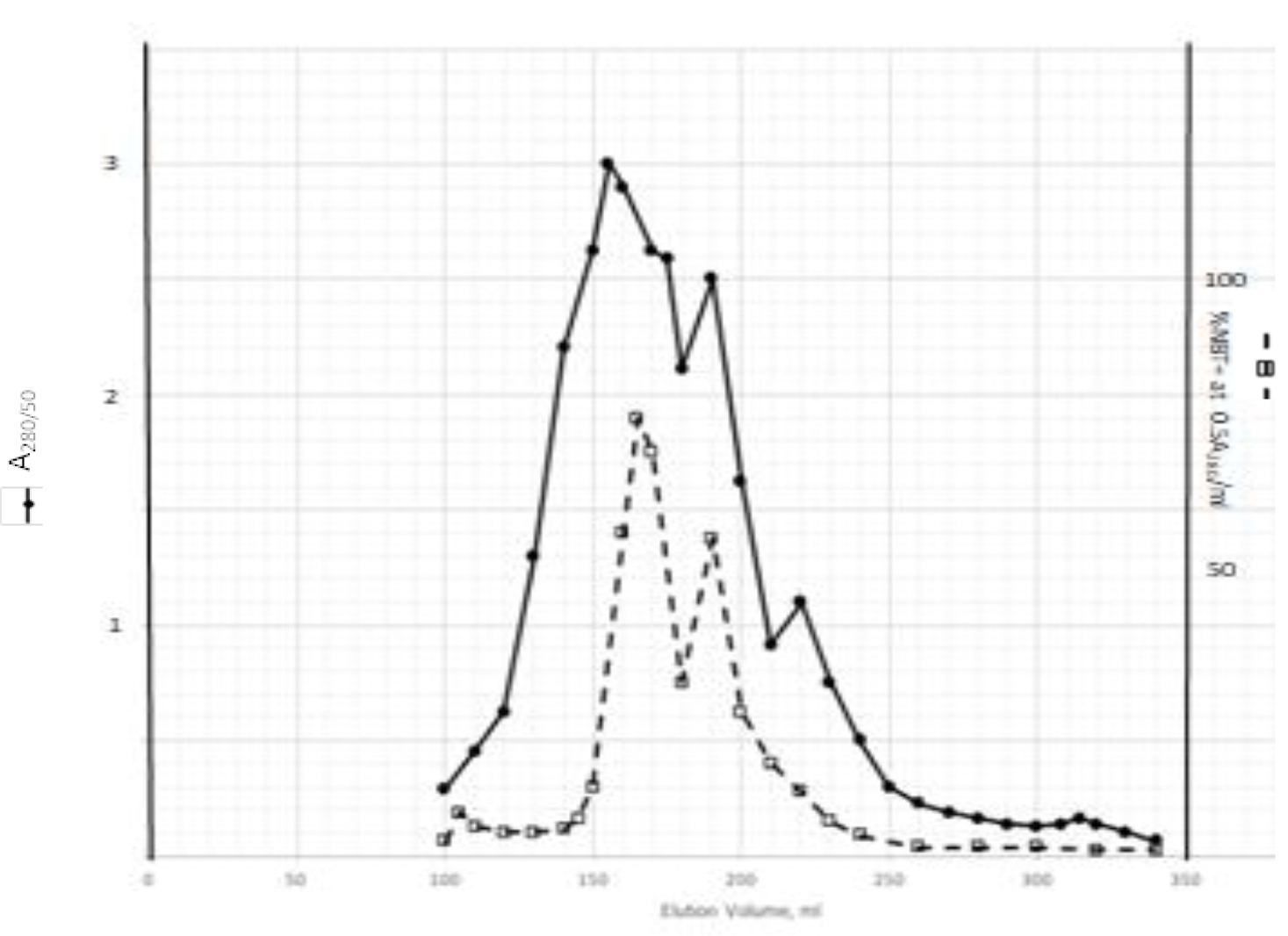
Figure 4. Sephadex LH20 chromatography of Kav=0.43 + 0.52 pooled material of Fig. 1
Kav=0.43 + 0.52 of Fig. 1 were recovered by C18 cartridges as described in Methods and Materials. The residue was dissolved in methanol. 4 ml of methanol solution containing approximately 25,000 A280 absorption units were put on a column of Sephadex LH20 for chromatography as described In Methods and Materials. Two fractions eluted between 150–180 ml and 180–210 ml were separately pooled, and methanol was removed by rotary evaporation under vacuum.
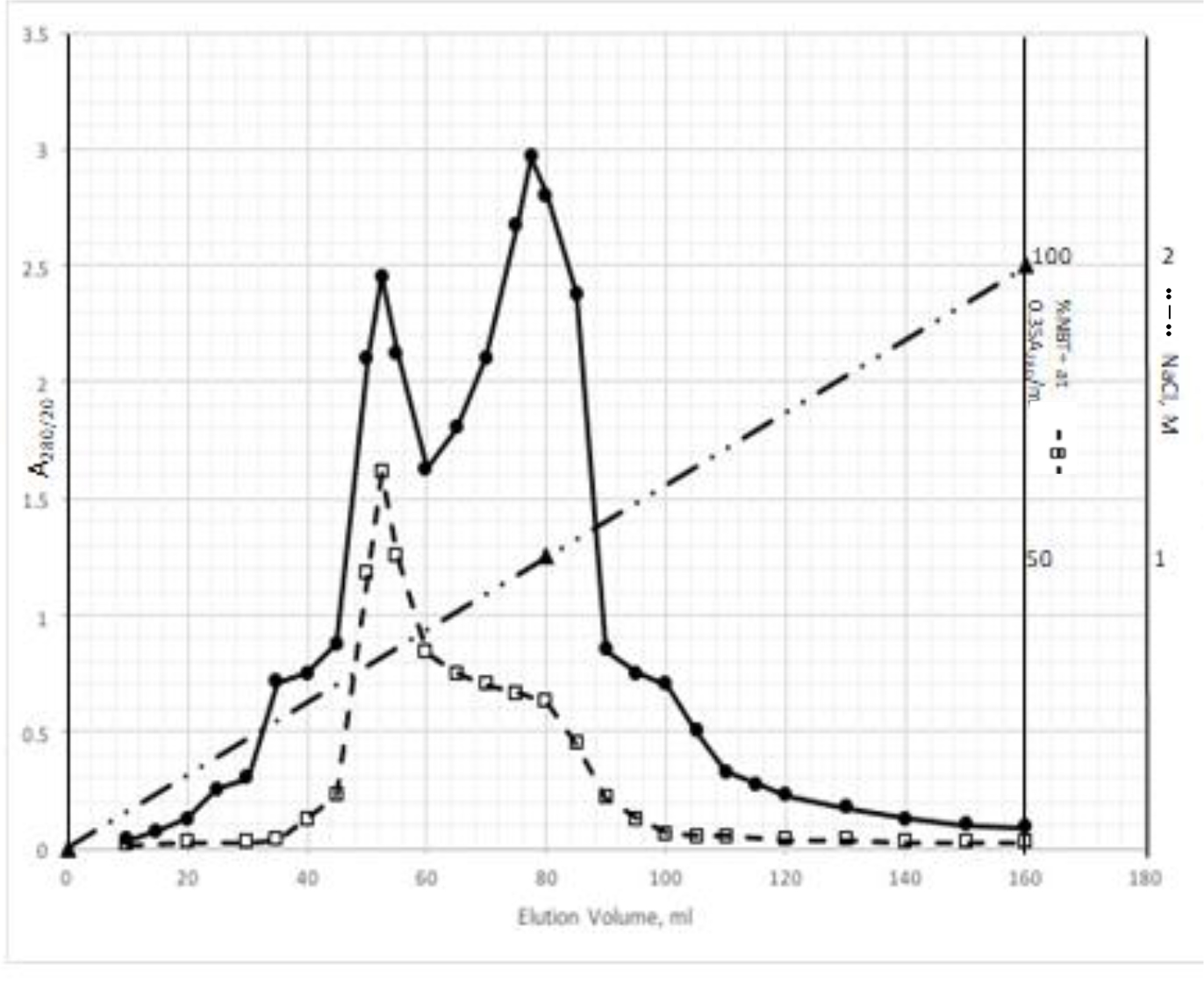
Figure 5. DEAE-Sephadex chromatography of the 150–180 ml peak of Sephadex LH20 chromatography
The residue of the 150–180 ml peak of Fig. 4 was dissolved in dilute NaOH, and pH adjusted to 7.8. 5 ml of this solution containing a total of 11,500 A280 absorption units were put onto a column of DEAE-Sephadex for chromatography as described in Methods and Materials. Active fractions eluted between 0.55–0.75 M NaCl and 0.85–1.05 M NaCl were separately pooled.
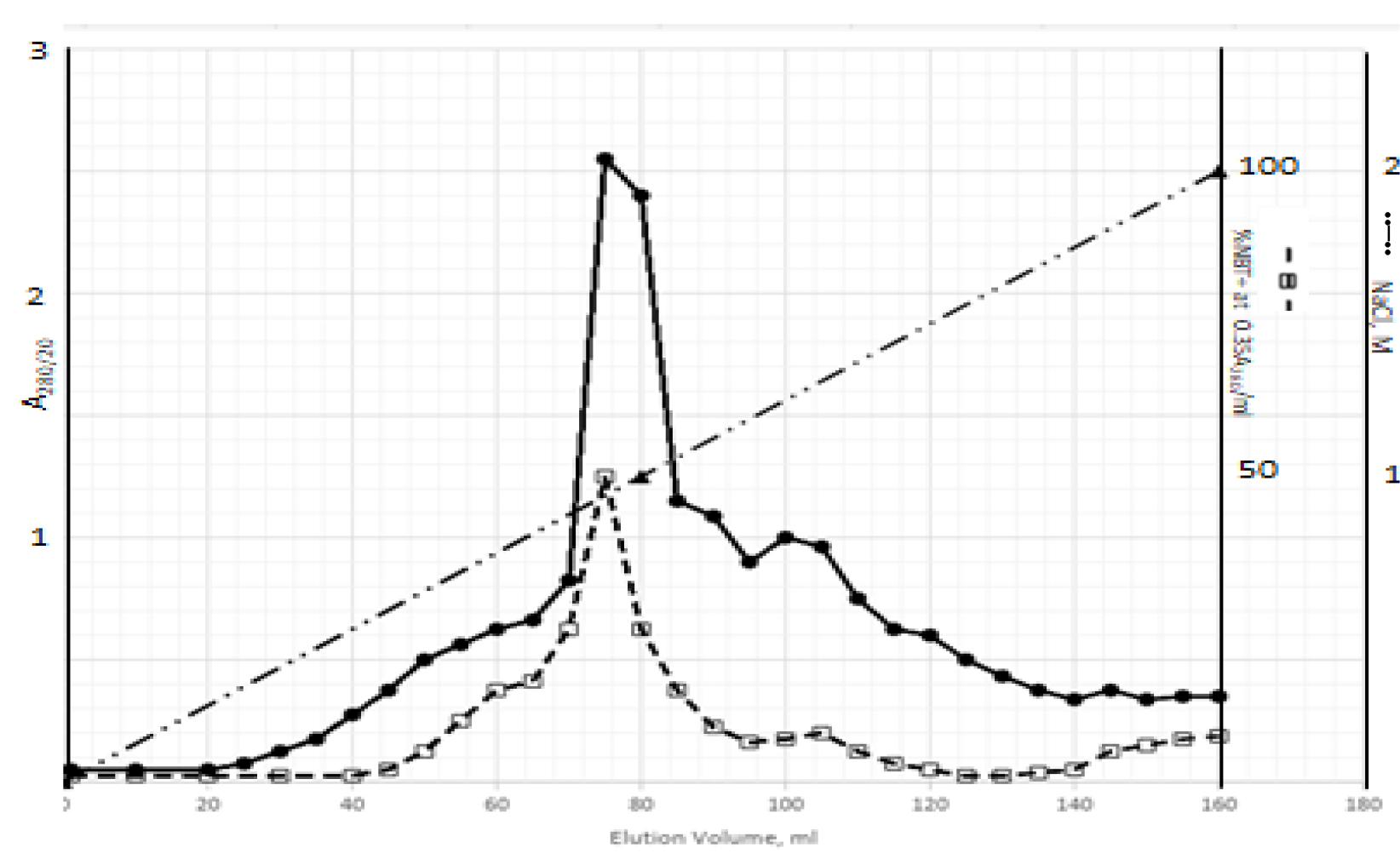
Figure 6. DEAE-Sephadex chromatography of the 180–210 ml peak of Sephadex LH20 chromatography
The residue of the 180–210 ml peak of Fig. 4 was dissolved in dilute NaOH, and pH adjusted to 7.8. 5 ml of this solution containing a total of 5,500 A280 absorption units were put onto a column of DEAE-Sephadex for chromatography as described in Methods and Materials. Active fractions eluted between 0.55–0.75 M NaCl and 0.85–1.05 M NaCl were separately pooled.
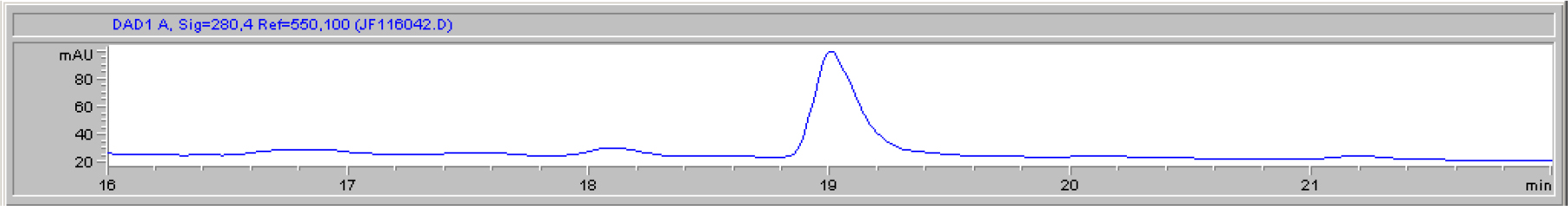
Figure 7. HPLC purification of the DHI dissociated from DI of CDA-2
The HPLC RT-19.1 peak was a major UV absorption peak of the precipitate from the eluant of 0.55–0.75 M NaCl of DEAE-Sephadex chromatography shown in Fig. 5. On final analysis, it yielded a single peak at RT-19.1. HPLC was carried out as described in Methods and Materials.
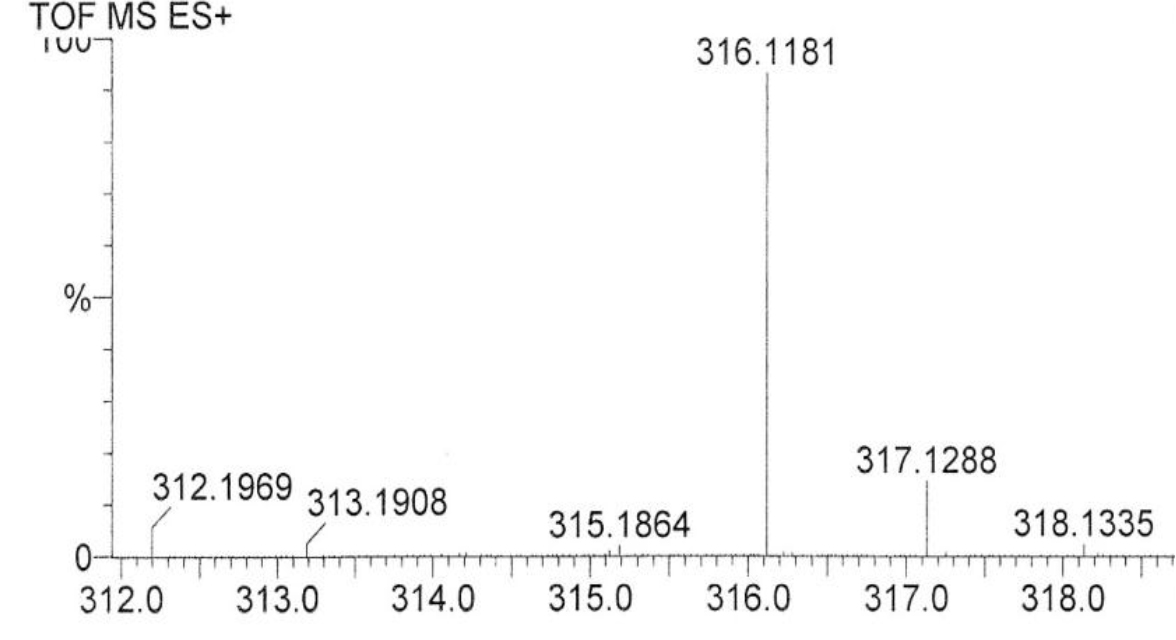
Figure 8. Mass spectroscopy of the RT-19.1 peak of HPLC
An aliquot of the methanol solution of the material purified to a single HPLC peak shown in Fig. 7 was injected into LC/MS instrument for the mass determination as described in Methods and Materials.
Confirmation of the HPLC RT-19.1 peak of CDA-2 as pregnenolone
Pregnenolone obtained from Sigma was dissolved in methanol to make 1.5 mg/ml, namely 4.74 mM. The A280 absorption is very low, which is 0.054/mM. The solution gave a HPLC peak at RT-19.1. When RT-19.1 peak of CDA-2 was mixed with Sigma pregnenolone, the mixture gave a single peak at RT-19.1. The potency of the HPLC RT-19.1 of CDA-2 as DHI was exactly the same as that of Sigma pregnenolone as previously reported, [7], namely 7.16 µM. It is obvious that pregnenolone is a component of active liposomal DIs of CDA-2.
Acidic pentapeptides of hemoglobin as DIs
We have tried to capture DIs in the supernatant of DEAE-Sephadex active eluants. The significant A280 absorption peaks of HPLC were all inactive as DI. We thus concluded that DIs of CDA-2 might not have A280 absorption. We have previously found that acidic peptides of the urine were active Dis [42]. We had reason to believe that plasma and urinary peptides were primarily the degradative products of endogenous proteins, because peptide profiles among different persons were similar, and those profiles were most close to the peptide profile of the spleen extract [40,41]. It is well known that erythrocytes are constantly turning over, and spleen is the organ to degrade dead erythrocytes. We assumed that the active acidic peptide of CDA-2 might be the degradative products of hemoglobin. Taking cue from the discovery of an acidic pentapeptide to modulate hemopoiesis by Laerum and Paukovits [78], we randomly picked pentapeptides containing at least two acidic amino acid residues from the sequences of α- and β-hemoglobin for synthesis to test their activities as DIs. Indeed, acidic pentapeptides of hemoglobin were active as DIs as shown in Table 2, although the activities were only modest.
Table 2. DI activity of synthetic acidic pentapeptides
|
Designation
|
Sequence
|
ED25, µM
|
|
5P-1
|
Ala-Glu-Ala-Leu-Glu
|
122±32
|
|
5P-2
|
Val-Asp-Glu-Val-Gly
|
154±28
|
|
5P-3
|
Val-Asp-Asp-Met-Pro
|
174±55
|
|
5P-4
|
Asp-Pro-Glu-Asp-Phe
|
207±42
|
|
PA-5P-4
|
PA-Asp-Pro-Glu-Asp-Phe
|
161±33
|
|
5P-5
|
Pro-Glu-Glu-Lys-Ser
|
415±76
|
Synthetic acidic pentapeptides were dissolved in 80% methanol for the test of DI activity. The dosages were selected to give ED between 20 and 30% NBT + . ED25 was obtained from the plots, and expressed as mean±S. D. from at least two experiments.
The combination of RA and acidic pentapeptides produced additive effect, whereas the combination of RA and DHIs produced synergistic effect as previously reported [7,31,76,77].
RA is a well known DI, which requires retinoic acid receptor {RAR} to activate oligoisoadenylate synthetase [79], and the product of this enzyme, oligoisoadenylate, is the responsible DI to initiate differentiation induction [6]. Oligoisoadenylate has to be synthesized inside the cell to function, because 5’ terminal triphosphate prohibits it to be absorbed from outside. Therefore, RA is only effective on cells expressing RAR. RARs are expressed on developmental stages of embryonic cells [80], in pluripotent stem cells to regulate organ and limb development [81], in CSCs [82–84], and in certain cancers such as APL [27], acute myeloid leukemia [85], neuroblastoma [86], breast cancer [87], and melanoma [88].
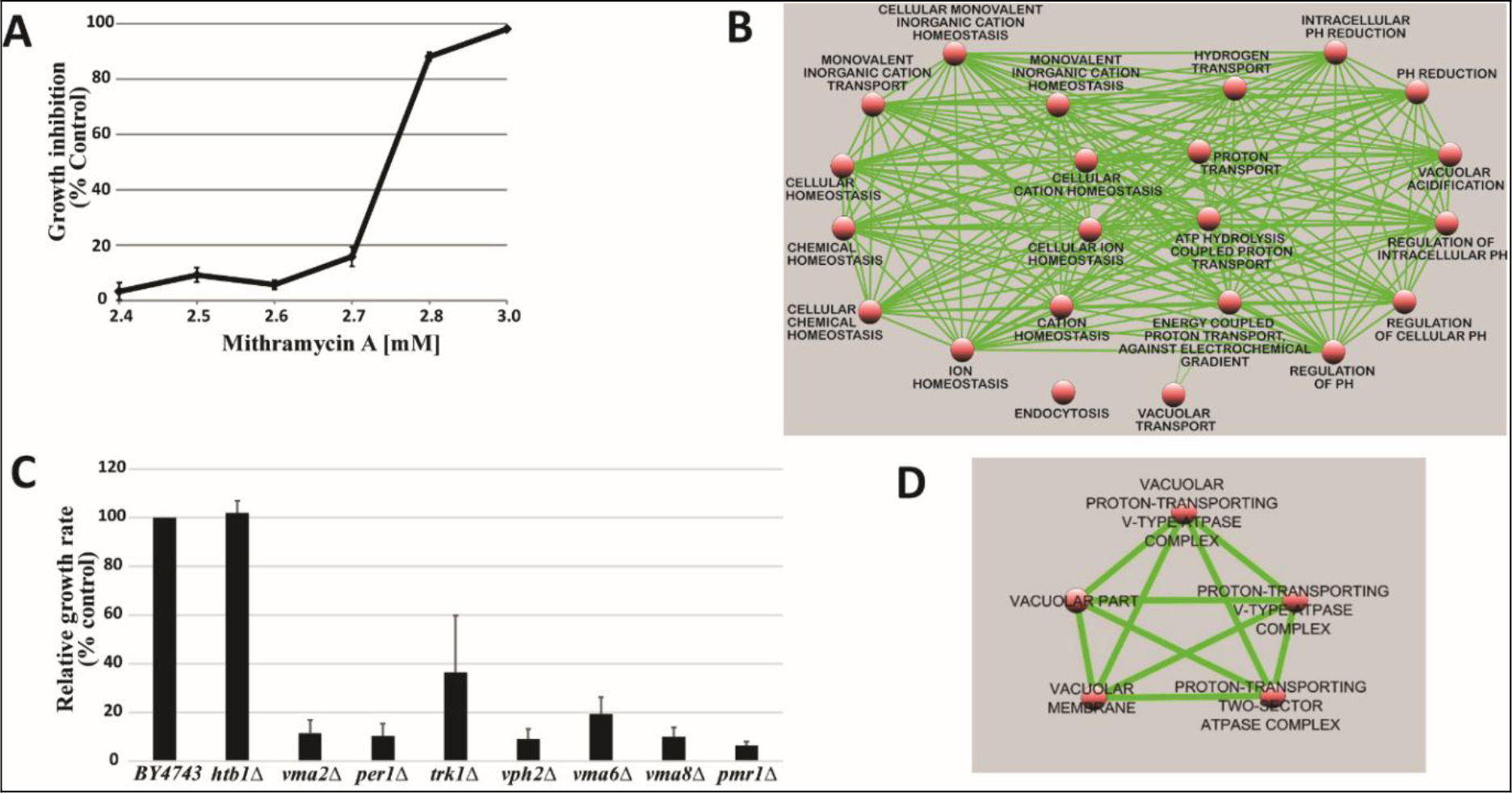
TPA is another well known DI, which acts as DI in some cells, but as differentiation inhibitor in other cells depending on which molecules are being affected, or as tumor promoter by increasing cell multiplication in initiated cells [89]. Biological effects of TPA are mediated through initial interaction with membrane [90]. Therefore, TPA affects almost all cancers. The tumor promotion activity of TPA can be effectively cancelled by DHIs [91]. Despite its adverse effects, TPA has been put through clinical trials for the evaluation as a cancer drug [92–94]. TPA had a very impressive DI activity on HL-60 cells as shown in Fig. 9. The maximum activity was at 0.4 nM to induce 92% NBT + . At this concentration 90% of cells were attached to the culture dish. Above this concentration, cell growth was greatly inhibited, and NBT + cells declined precipitously. ED25 of TPA was 0.17 ± 0.02 nM when tested on fast growing HL-60 cells. The growth rates measured by N3/N0 were between 13.7–15.4, almost 3 to 4 generations in 3 days. ED25 of RA was 1.17 ± 0.16 µM tested on HL-60 cells with growth rates same as those of TPA. We have previously found that HL-60 cells responded better to RA when grew slower at earlier passages. ED25 of RA was 0.78 µM when tested on HL-60 cells with N3/N0 between 6.3–7.5.
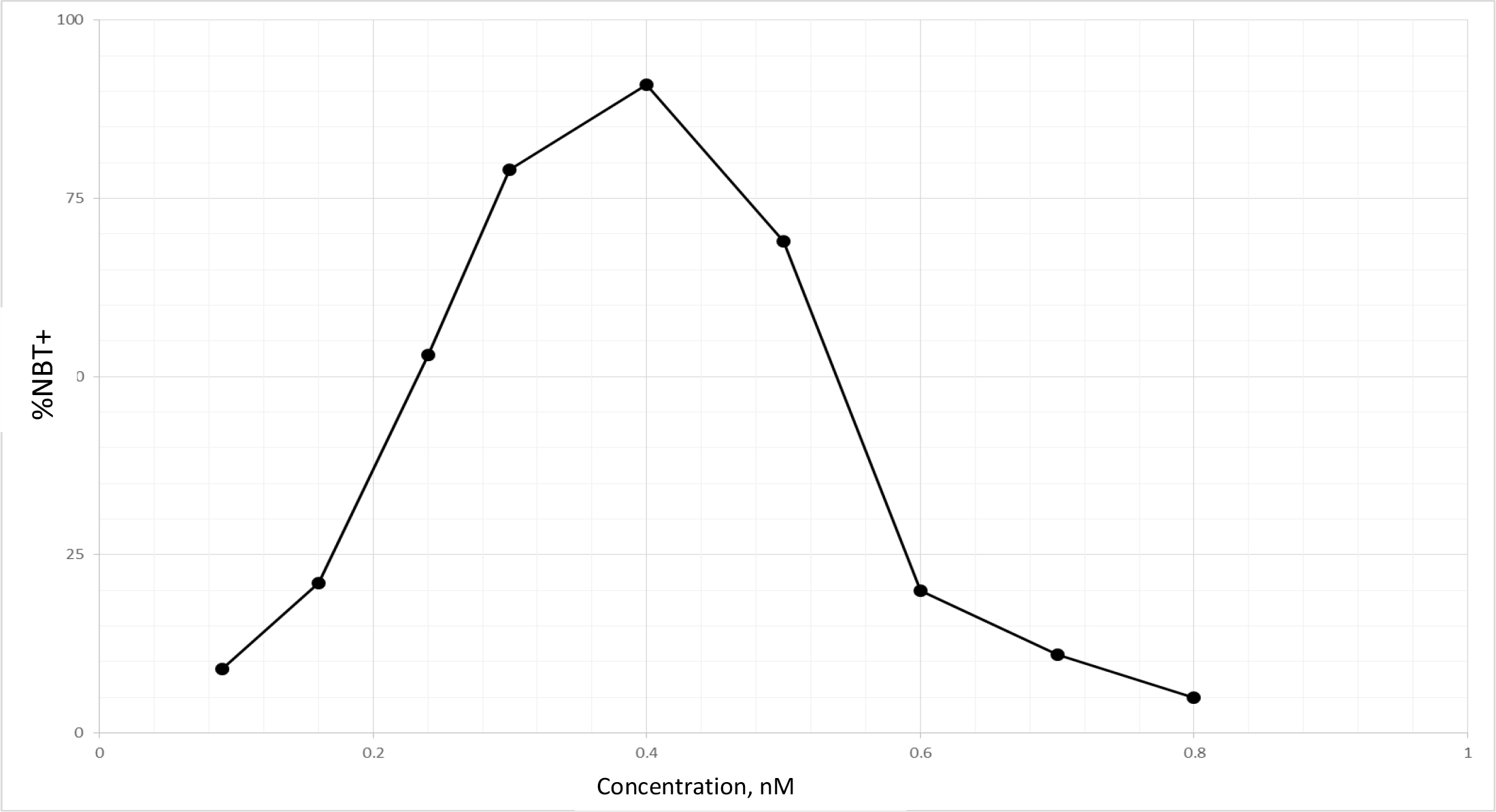
Figure 9. DI activity of TPA
TPA was dissolved in methanol which was added to the culture in the amounts indicated. The volume of methanol was less than 2%. NBT assay was conducted as described in Methods and Materials.
Potential DHIs for synthetic CDA formulations
We have previously shown that RA alone could not push all HL-60 cells to complete TD [7]. 95% NBT + cells was the maximum extent achieved by RA. But in the presence of DHIs, the extent of NBT + cells could reach 100% [7]. 92% NBT + cells was the maximum extent reached by TPA in this study. Cells unable to undergo differentiation in the presence of DI alone are the cells damaged enough to prevent completion of two replication cycles needed for TD. The damaged cells if repaired become the roots for recurrence. Removal of the possible recurrent roots by DHIs is another important contribution of DHIs. The most serious damage that can be attributed to DIs are the promotion of the dissociation of methylation enzymes, and the conversion of the monomeric MTs into nucleases. MT inhibitors can effectively prevent such conversion. Uroerythrin was a good MT inhibitor to function as the most active DHI of CDA-2 [77]. This chemical is not commercially available. We need to find substitutes to prevent damages created by DIs alone. We have previously found that I3 and I4 from limited alkaline hydrolysis of poly I were potent inhibitors of nucleolar rRNA methyltransferase [2]. We, thus, prepared I3 + I4 and A3 + A4 as previously described [2] to test their DHI activities. Results shown in Table 3 indicate that I3 + I4 and A3 + A4 are active as DHIs. Included in Table 3 is PP as the most active DHI we have discovered so far. PP has been shown active as signal transduction inhibitor and inhibitor of androgen receptor,[95–98]. Both classes of inhibitors are excellent DHIs [7]. The superb activity of PP as DHI may be attributable to its dual activities to inhibit signal transduction and steroid receptor.
Table 3. Potential DHIs for synthetic CDA formulations
|
DHI
|
RI0.5
|
|
I3+I4
|
3.7±0.58 µM
|
|
A3+A4
|
4.8±1.12 µM
|
|
PP
|
12.2±2.2 nM
|
10 mg of poly I and poly A was dissolved in 1 ml of 0.3N KOH and incubated at 37o C for 0.5 h. The solution was neutralized with HCl, and put onto a column of DEAE-Sephadex for chromatography as previously described,[2]. I3 and I4 in the ratio of 1:0.9 were the major components, which were combined and recovered as previously described,[2] for the test of DHI activity. The preparations of oligonucleotides were dissolved in 80% methanol, and pp was dissolved in methanol for the determination of RI0.5 as previously described,[7]. HL-60 cells for these experiments had growth rates of N3/N0 between 7.5–9.8.
Development of synthetic CDA formulations for the prevention and therapy of cancer via targeting of CSCs
CDA-2 was effective to eliminate CSCs subpopulation [99,100], and had a very effective therapeutic efficacy on MDS [62, 63], a disease attributable to CSCs. Our immediate objective is to develop synthetic CDA formulations for the therapy of MDS, CSCs, and cancers in which CSCs play a dominant role on the pathological features and drug responses of the diseases. Cancers originated from progenitor stem cells such as MDS above described and acute myeloid leukemia resulting from MDS are enriched in CSCs. Evidence indicates that brain tumors are also originated from progenitor stem cells [101,102]. Melanoma and pancreatic cancer may not originate from progenitor stem cells, but these cancers express high levels of factors such as hypoxia inducible factors and NF-kB that can effectively drive EMT to display CSC phenotype [103 -106]. On MDS, protection of pathological cells is a necessity, because the therapy requires the differentiation of pathological cells to become functional erythrocytes. DIs and DHIs of low toxicity are preferred. On other CDA formulations, destruction of pathological cells is a preferred choice. On CSCs, toxicity is also a big concern, because CSCs naturally reject toxic chemicals. CSCs are situated in hypoxic microenvironment hard to reach by the blood, small molecules with penetration power are preferred. On brain tumor, blood brain barrier must be the major concern. Hydrophobic chemicals have better chance to cross that barrier. For melanoma and pancreatic cancer, potent DIs and DHIs are preferred. CDA formulations must consist of both DIs and DHIs to make perfect drugs. The following formulations are acceptable formulations: 3xED25 of DI + 1xRI0.5 of DHI; 2xED25 of DI + 2xRI0.5 of DHI; or 1xED25 of DI + 3xRI0.5 of DHI. 1xRI0.5 of DHI is equivalent to 1xED25 of DI [7]. The multiplicity of DI can be multiplicity of a single DI, or a combination of different DIs. Likewise, the multiplicity of DHI can be multiplicity of a single DHI, or a combination of different DHIs.
Our deliberated CDA formulations are listed in table 4. CDA-MDS, CDA-BT (brain tumor), and CDA-M&P (melanoma and pancreatic cancer) are parenteral preparations, and CDA-CSC is an oral preparation which is designed for long term application to prevent recurrence after active therapy. Thus, an oral preparation is a practical drug form. On active cancer therapy, CDA-CSC is best used in combination with drugs to inhibit growth factor receptors and signal transductions, or drugs for anti-steroid hormonal therapy. These drugs are actually good DHIs. It is also compatible with other therapeutic modalities to target on different entities, e.g. CDA-CSC to target on CSCs and cytotoxic drugs or radiation to target on non-CSCs. TPA and PP are not soluble in H2O. RA, resveratrol, and curcumin are not very soluble in H2O. Dispensing aids such as liposomal and nanoparticle technologies must be employed to increase bioavailability for clinical application. Phenylacetylglutasmine {PAG} is a major chemical constituent of CDA-2, which is also very useful to extend bioavailability of DIs and DHIs. We recommend to include 2 mM PAG in each CDA formulation for the purpose of extending bioavailability of DIs and DHIs and to cancel tumor promotion activity of TPA. We have previously shown that PAG, namely Antineoplaston A10, was effective to prevent the loss of low molecular weight metabolites including active DIs and DHIs [40]. By preventing the loss of endogenous DIs and DHIs, PAG was effective to prevent carcinogens-induced pulmonary neoplasia and hepatoma [107,108], and to achieve therapy of early stage cancer [40]. PAG is a very effective deterrent of tumor promoters. PAG is synthesized from phenylacetyl chloride and glutamine as previously described [76]. CDA formulations listed in Table 4 are plasma concentrations to achieve induction of TD of HL-60 cells above 100% on purpose to achieve maximum therapeutic effect. Multiplication of these amounts per liter of blood by a factor of 5, which is the normal blood volume of a person of 80 Kg body weight, is necessary to produce an effective dosage. Application of 3 effective dosages a day should give satisfactory therapy.
Table 4. Synthetic CDA formulations
|
|
Effect on HL-60 cells
|
|
Designation
|
Formulation
|
% Growth
|
%NBT+
|
|
CDA-MDS
|
RA(ED25)-5P-1(ED25)-I3+I4(RI0.5)-PP(RI0.5)-Na pregnenolone SO4(RI0.5)
|
77 ± 5.2
|
100
|
|
CDA-CSC
|
RA(ED25)-TPA(ED25)-PP(RI0.5)-resveratrol(RI0.5)-curcumin(RI0.5)
|
53 ± 3.6
|
100
|
|
CDA-BT
|
TPA(2xED25)-PP(2xRI0.5)-Na phenylbutyrate (RI0.5)-pyrogallol(RI0.5)
|
45 ± 3.9
|
100
|
|
CDA-M&P
|
TPA(2xED25)-5P-1(ED25)-PP(2xRI0.5)-Na tannate (RI0.5)
|
33±4.4
|
100
|
CDA-MDS is a parenteral preparation as a monotherapy of MDS. CDA-CSC is an oral preparation for use in combination with other drugs. CDA-BT is a parenteral preparation as a monotherapy of brain tumor. CDA-M&P is also a parenteral preparation as a monotherapy for melanoma and pancreatic cancer.
DIs and DHIs were methanol solution except 5P-1 which was in 80% methanol, and Na phenylbutyrate which was in H2O solution. Each component was added individually. RA(ED25) is 1.17 µM, 5P-1(ED25) is 122 µM, I3+I4(RI0.5) is 3.7 µM, PP(RI0.5) is 12.2 nM, TPA(ED25) is 0.17 nM, resveratrol (RI0.5) is 1.16 µM, curcumin(RI0.5) is 1.24 µM, Na phenylbutyrate(RI0.5) is 2 mM, and Na tannate(RI0.5) is 0.37 µM as previously reported,[7] or shown in this paper. NBT assay was carried out as described in Methods and Materials.
Discussion
Abnormal methylation enzymes are an important issue of cancer. Almost all human cancers display such an abnormality [8,109]. Abnormal methylation enzymes are the critical factor responsible for the blockade of differentiation of cancer cells [6]. The expression of telomerase fits the first hit of Knudson’s two hits theory [110]. Telomerase is also expressed in primitive stem cells such as embryonic stem cells and progenitor stem cells. Evidently blockade of differentiation is a normal biological process to build up cell mass for the development of fetus, or wound healing. The blockade of differentiation in these normal stem cells does not create problem, because Tet enzymes are functioning which can break through the blockade created by abnormal methylation enzymes to spearhead differentiation programs. The problem arises if Tet enzymes become dysfunction due to mutation or silencing such as the case of MDS [23–26]. The loss of Tet enzymes becomes the second hit of Knudson’s theory. Therefore, the evolution of cancer in the case of MDS is a perfect interpretation on the validity of the two hits theory of Knudson [110], and our chemosurveillance hypothesis [40].
Progenitor stem cells and CSCs are almost indistinguishable with only very minor differences, the functionality of Tet enzymes being a significant difference between them. Progenitor stem cells are very likely the origin of most human cancers, not just acute myeloid leukemia resulting from MDS or brain tumors. We have detected abnormal methylation enzymes to function actively in preneoplastic hyperplastic nodules induced by hepatocarcinogen [111]. Therefore, progenitor stem cells are just like time bombs, and natural DIs and DHIs are the safety devices to keep them from evolving into CSCs.
Chemicals extractable by dichloromethane are in general very hydrophobic. The detection of pregnenolone as a component of DIs of CDA-2 is a certain indication that active DIs of CDA-2 is liposomal complexes. The release of liposomal-like DIs from methanol extractable DIs by NaOH hydrolysis suggests that dichloromethane extractable DIs and methanol extractable DIs are basically the same liposomal complexes. A large proportion of these liposomal complexes become covalently linked to the inactive carrier previously identified as pigment peptides [40] either by enzymatic dehydration in the body or due to heat pyrolysis during evaporation of ethanol eluant in the process of CDA-2 preparation. The inactive pigment peptide carrier is very likely fragments of membrane. The liposomal DIs of Kav = 0.43 and 0.52 are stable in methanol, ethanol, and dichloromethane, but are not stable in butanol or high slat solution. DIs in free forms do not seem to have A280 absorption. Without A280 absorption as a probe, it is very difficult to capture DIs of CDA-2. The real DIs of CDA-2 are still up in the air.
We have identified pregnenolone as a DHI of CDA-2 in this study. Pregnenolone is the master substrate of all biologically active steroids, which is synthesized from cholesterol absorbed from diet or produced in the liver. The peak age of the production of pregnenolone is 20 years old, producing approximately 50 mg a day,[112]. The very young and the very old are the two age groups producing the least amount of pregnenolone. These are the two age groups most susceptible to cancer. Fortifying effective DHIs definitely is a good policy to amend natural insufficiency.
The therapy of APL with RA sets a classical example of the effectiveness of destabilization of abnormal methylation enzymes on cancer therapy [27]. RA alone is very effective, but imperfect. Recurrence ensues quickly [28]. This is because RA alone causes the dissociation of abnormal methylation enzymes too extensive to contribute to nuclease activity which creates damages to interrupt differentiation process. This phenomenon is analogous to the antiviral effect of interferon. Interferon is actually a DI like TPA to activate oligoisoadenylate synthetase through interaction with membrane. The product of oligoisoadenylate synthetase is a potent activator of latent nucleases to execute antiviral effect. The damage created by DI alone may affect only a small fraction of cells in the S phase when DNA is most susceptible to nuclease attack. The damages interrupt differentiation process, but when damaged cells are repaired later they become the roots for recurrence. Damages by DI alone can be prevented by DHIs. MT inhibitors can prevent protease modification to turn MT into nuclease. Steroids analogs are even better, which keep MT in dimeric complex with SAHH to protect the integrity of MT. The protection of MT from becoming nuclease is the cause that DHI is an essential partner of a perfect cancer drug.
The option for the eradication of cancer stem cells is very limited. The ideal therapeutic agents must be small molecules that are relatively non-toxic to bypass drug afflux pumps to reach adequate intracellular concentrations to trigger cancer stem cells to undergo differentiation. Synthetic CDA formulations are an attractive strategy to eradicate cancer stem cells. DIs is the most important components of CDA formulations. The choices of DIs are very limited; RA is only effective in cancers expressing RAR. TPA is universal, but is very toxic. 5Ps are universal, but are not very active. The requirement of DI is to initiate TD to reach a level above 15%, the rest can be accomplished by DHIs [7]. Therefore, even though 5Ps are not very active, they can be remedied by very active DHIs such as PP. We have developed many excellent DHIs to choose from. The mere application of effective DHIs can have excellent therapeutic effect on cancer as exemplified by imatinib mesylate. The brain compartment is a unique compartment protected by blood brain barrier, which also protect the loss of endogenous metabolites. Therefore, the deficiency of endogenous DIs and DHIs is not as severe as other body compartments. That is why DHIs have impressive therapeutic effect on brain tumor.
Destabilization of abnormal methylation enzymes is a good policy to implement at the earliest diagnosis of cancer to avoid aberrant DNA methylation no matter what cancer therapy is to follow. Aberrant DNA methylations are frequently detected in advanced cancer, which can turn ordinary responsive cancer to become very vicious cancer unresponsive to any treatment. Aberrant DNA methylations happen more readily when DNA synthesis is slowing down by cytotoxic drugs or radiation [113]. Another benefit to implement destabilization of abnormal methylation enzymes is to avoid expansion of CSC population induced by the destruction of the tumor caused by cytotoxic chemotherapy [114], or due to tumor progression [103–106]. Reconstruction of the tumor is a major mission of CSCs. Therefore, destabilization of abnormal methylation enzymes is a very good strategy to pursue for cancer therapy.
Acknowledgement
This study was supported in part by a contract awarded to CDA Therapeutics, Inc. by Xinhua Pharmaceutical Company of Zibo, Shandong China. We are indebted to the gift of CDA-2 solution from Mr. Zanji Sun of NTPharma of Tai Zhou, Jiangsu, China. We are very grateful to Christian Browder for the art work of figures.
Funding Information
The funder had no involvement in the study design; in the collection, analysis and interpretation of the data; in the writing of the report, and in the decision to submit the paper for publication. The funder had an active supervising role.
References
- Racanelli AC, Turner FB, Kie TY, Taylor SM (2008) A mouse gene that coordinates epigenetic controls and transcriptional interference to achieve tissue-specific expression. Mol Cell Biol 281: 836–848.
- Liau MC, Hunt ME, Hurlbert RB (1976) Role of ribosomal RNA methylases in the regulation of ribosome production in mammalian cells. Biochemistry 15: 3158–3164. [crossref]
- Bernstein KA, Bleichert F, Bean JM, Cross FR, Baserga SJ (2007) Ribosome biogenesis is sensed at the Start cell cycle checkpoint. Mol Biol Cell 18: 953–964. [crossref]
- Justilien V, Ali SA, Jamieson L, Yin N, Cox AD, et al. (2017) Ect2-Dependent rRNA Synthesis Is Required for KRAS-TRP53-Driven Lung Adenocarcinoma. Cancer Cell 31: 256–269. [crossref]
- Liau MC, Chang CF, Saunders GF, Tsai YH (1981) S-Adenosylhomocysteine hydrolases as the primary target enzymes in androgen regulation of methylation complexes. Arch Biochem Biophys 208: 261–272.
- Liau MC (2004) Abnormal methylation enzymes: a selective molecular target for differentiation therapy of cancer. Chin Pharm J 56: 57–67.
- Liau MC, Kim JH, Fruehauf JP (2019) Potentiation of ATRA activity in HL-60 cells by targeting methylation enzymes. Pharmacol Pharmaceut Pharmacovig 3: 9–17
- Liau MC, Zhuang P, Chiou GCP (2010) Identification of the tumor factor of abnormal methylation enzymes as the catalytic subunit of telomerase. Chin Oncol Cancer Res 7: 86–96.
- Prudova A, Bauman Z, Braun A, Vitvitsky V, Lu SC, et al. (2006) S-adenosylmethionine stabilizes cystathionine beta-synthase and modulates redox capacity. Proc Natl Acad Sci USA 103: 6489–6494. [crossref]
- Chiba P, Wallner L, Kaizer E (1988) S-Adenosylmethionine metabolism in HL-60 cells: effect of cell cycle and differentiation. Biochim Biophys Acta 971: 38–45.
- Liau MC, Lee SS, Burzynski SR (1989) Hypomethylation of nucleic acids: a key to the induction of terminal differentiation. Intl J Exp Clin Chemother 2: 187–199.
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, et al. (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324: 930–935. [crossref]
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, et al. (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333: 1300–1303. [crossref]
- Shen L, Song CX, He C, Zhang Y (2014) Mechanism and function of oxidative reversal of DNA and RNA methylation. Annu Rev Biochem 83: 585–614. [crossref]
- Wu X (2017) TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet 18: 517–534. [crossref]
- He YF, Li BZ, Li Z, Liu P, Wang Y, et al. (2011) Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333: 1303–1307. [crossref]
- Maiti A, Drahat AC (2011) Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implication for active demethylation of CpG sites. J Biol Chem 286: 35334–35338.
- Hu L, Lu J, Cheng J, Rao Q (2015) Structural insight into substrate preference for TET-medicated oxidation. Nature 527: 118–122.
- Huang Y, Charez L, Chang X, Wang X (2014) Distinct role of the methylcytosine oxidases Tet 1 and Tel 2 in mouse embryonic stem cells. Proc Natl Acad Sci USA 111: 1361–1366.
- Iqbal K, Jin SG, Pfeifer GP, Szabó PE (2011) Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc Natl Acad Sci U S A 108: 3642–3647. [crossref]
- Ficz G, Branco MR, Seisenberger S, Santos F (2011) Dynamic regulation of 5-hydroxymethylcytosine mouse ES cells and during differentiation. Nature 473: 398–403.
- Monfort A, Wutz A (2013) Breathing-in epigenetic change with vitamin C. EMBO Rep 14: 337–346. [crossref]
- Haffiner MC, Chaux A, Meeker AK, Esopi D (2011) Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget 2: 627–637.
- Kudo Y, Tateishi K, Yamamoto K, Yamamoto S (2012) Loss of 5-hydroxymethylcytosine is accompanied with malignant cellular transformation. Cancer Sci 103: 670–676.
- Ficz G, Gribben JG (2014) Loss of 5-hydroxymethylcytosine in cancer: cause or consequence? Genomics 104: 352–357. [crossref]
- Wang KC, Kang CH, Tsai CY, Chou NH (2018) Ten-eleven translocation 1 dysfunction reduces 5-hydroxylmethylcytosine expression levels in gastric cancer cells. Oncol Lett 15: 278–284.
- Huang M, Ye Y, Chen S, Chai JR (1988) Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 72: 567–572.
- Warrell RP, Frankel SR, Miller MA, Scheinberg DA (1991) Differentiation therapy of acute promyelocytic leukemia with tretinoin (all-trans retinoic acid). N Engl J Med 304: 1385–1393.
- de Thé H, Pandolfi PP, Chen Z (2017) Acute Promyelocytic Leukemia: A Paradigm for Oncoprotein-Targeted Cure. Cancer Cell 32: 552–560. [crossref]
- Gambacorti-Passerini C, Antolini L, Mahon FX, Guilhot F, Deininger M, et al. (2011) Multicenter independent assessment of outcomes in chronic myeloid leukemia patients treated with imatinib. J Natl Cancer Inst 103: 553–561. [crossref]
- Liau MC, Liau CP, Burzynski SR (1992) Potentiation of induced terminal differentiation by phenylacetic acid and related chemicals. Intl J Exptl Clin Chemother 5: 9–17.
- Baker MJ, Brem S, Deniels S, Sherman B (2002) Complete response of a recurrent, multicentric malignant glioma in a patient treated with phenylbutyrate. J Neurooncol 59: 239–242.
- Burzynski SR, Nagy-Kubove E (2011) Treatment of esthesioneuroblastoma and non-small cell-lung cancer with phenylbutyrate. J Cancer Ther 2: 518–522.
- Burzynski SR, Tamicki TJ, Burzynski GS, Brookman S (2014) Preliminary findings on the use of targeted therapy with pazopanib and other agents in combination with sodium phenylbutyrate in the treatment of glioblastoma multiforme. J Cancer Ther 5: 1423–1437.
- Burzynski SR, Burzynski GS, Brookman S (2015) A case of sustained objective response of recurrent/progressive diffuse intrinsic pontine glioma with phenylbutyrate and targeted agents. J Cancer Ther 6: 40–44.
- Stoner GD, Wang LS, Zikri N, Chen T, Hecht SS, et al. (2007) Cancer prevention with freeze-dried berries and berry components. Semin Cancer Biol 17: 403–410. [crossref]
- Amin AR, Kucuk O, Khuri FR, Shin DM (2009) Perspectives for cancer prevention with natural compounds. J Clin Oncol 27: 2712–2725. [crossref]
- Wang LS, Hecht SS, Carmella SG, Yu N (2009) Anthocyannin in black raspberries prevent esophageal tumors in rats. Cancer Prev Res 2: 84–93.
- Allegra A, Innao V, Russo S, Gorace D (2017) Anticancer activity of curcumin and its analogues: preclinical and clinical studies. Cancer Invest 35: 1–22.
- Liau MC, Szopa M, Burzynski B, Burzinski SR (1987) Chemosurveillance: a novel concept of the natural defense mechanism against cancer. Drug Exptl Clin Res 13: 77–82.
- Liau MC, Szopa M, Burzynski B, Burzinski SR (1987) Quantitative assay of plasma and urinary peptides as an aid for the evaluation of cancer patients undergoing Antineoplaston therapy. Drug Exptl Clin Res 13: 61–70.
- Liau MC, Lee SS, Burzynski SR. (1988) Differentiaiton inducing components of Antineoplaston A5. Adv Exptl Clin Chemother 6/88: 9–26.
- Liau MC, Lee SS, Burzynski SR (1990) Modulation of cancer methylation complex isozymes as a decisive factor in the induction of terminal differentiation mediated by Antineoplaston A5. Intl J Tiss React 12: 29–36.
- Williamson RJ, Kruger AR, Reynolds RJ (1994) Establishing the incidence of myelodysplastic syndromes. Br J Haemato 83: 743–748.
- Boula A, Voulgarelis M, Giannouli S, Katrinakis G (2006) Effect of CA2 antitumor necrosis factor-a antibody therapy on hematopoiesis of patients with myelodysplastic syndromes. Clin Cancer Res 12: 3099–3108.
- Itkin T, Rafii S (2017) Leukemic Cells “Gas Up” Leaky Bone Marrow Blood Vessels. Cancer Cell 32: 276–278. [crossref]
- Passaro D, Tullio AD, Abarrategi A, Rouault-Pierre K (2017) Increased vascular permeability in the bone marrow microenvironment contributes to disease progression and drug response in acute myeloid leukemia. Cancer Cell 32: 324–341.
- Counter CM, Mupta J, Harley CB (1995) Telomerase activity in normal leukocytes and hematological malignancies. Blood 85: 2315–2320.
- Fu C, Chen Z (2002) Telomerase activity in myelodysplastic syndrome. Chin Med J (Engl) 115: 1475–1478. [crossref]
- Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, et al. (2009) Mutation in TET2 in myeloid cancers. N Engl J Med 360: 2289–2301. [crossref]
- Lasson Cam Cote G, Quintas-Cardama A (2013) The changing mutational landscape of acute myeloid leukemia and myelodysplastic syndrome. Mol Cancer Res 11: 815–827.
- Papaemmanuil E, Gerstuug M, Malcovati L, Tauro S (2013) Clinical and Biological implication of driver mutations in myelodysplastic syndromes. Blood 122: 3616–3627.
- Bejar R, Lord R, Stovenson K, Bar-Natan M (2014) TET2 mutations predict response to hypomethylating agents in myelodysplstic syndrome patient. Blood 124: 2705–2712.
- Kennedy JA, Ebert BL (2017) Clinical implication of genetic mutations in myelodysplastic syndrome. J Clin Oncol 35: 968–974.
- Woll PS, Kjällquist U, Chowdhury O, Doolittle H, Wedge DC, et al. (2014) Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo. Cancer Cell 25: 794–808. [crossref]
- Maeck L, Haase D, Schoch C, Hiddemann W (2000) Genetic instability in myelodysplastic syndrome: detection of microsatellite instability and loss of heterozygosity in bone marrow sample with karyotype alteration. Br J Haemato 109: 842–846.
- Xie D, Hefmann KW, Mori N, Miller CW (2001) Allotype analysis of the myelodysplastic syndrome. Leukemia 14: 805–810.
- Kumamoto K, Ban S, Oda K, Tanaka H (2002) Chromosomal instability and radiosensitivity in myelodysplastic syndrome. Leukemia 16: 2253–2258.
- Takeda A, Goolsby C, Yaseen NR (2006) NUP98-HOXA9 induces long-term proliferation and blocks differentiation of primary human CD34+ hematopoietic cells. Cancer Res 66: 6628–6637. [crossref]
- Feng F, Li Q, Ling C, Zhang Y (2005) Phase III clinical trials of the cell differentiation agent-2 (CDA-2): therapeutic efficacy on breast cancer, non-small- cell lung cancer, and primary hepatoma. Chin J Clin Oncol 2: 706–716.
- Santi DV, Norment A, Garrett CE (1984) Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc Natl Acad Sci U S A 81: 6993–6997. [crossref]
- Ma J (2007) Differentiation therapy of malignant tumor and leukemia. CSCO Treaties on the Education of Clinical Oncology 480–486.
- Liau MC, Fruehauf JP (2015) Destabilization of abnormal methylation enzymes as a critical mechanism for CDA-2 to reverse MDS progression. The First International Forum of Myelodysplastic Syndrome in Tai Zhou, Jiangsu, China: 31–35.
- Prasanna P, Shack S, Wilson VL, Samid D (1995) Phenylacetate in chemoprevention: in vitro and in vivo suppression of 5-aza-2’-deoxycytidine-induced carcinogenesis. Clin Cancer Res 1: 865–871. [crossref]
- Jordan CT, Guzman ML, Noble M (2006) Cancer stem cells. N Engl J Med 355: 1253–1261. [crossref]
- Vermeulen L, Sprick MR, Kemper K, Stassi G, Medema JP (2008) Cancer stem cells–old concepts, new insights. Cell Death Differ 15: 947–958. [crossref]
- Zabierowski SE, Herlyn M (2008) Melanoma stem cells: the dark seed of melanoma. J Clin Oncol 26: 2890–2894. [crossref]
- Hemmings C (2010) The elaboration of a critical framework for understanding cancer: the cancer stem cell hypothesis. Pathology 42: 105–112. [crossref]
- Magee JA, Piskounova E, Morrison SJ (2012) Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell 21: 283–296. [crossref]
- Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, et al. (2001) The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med 7: 1028–1034. [crossref]
- Zhou M, Atkinson KL, Rosen JM (2010) Selective targeting of radiation resistant tumor initiating cells. Proc Natl Acad Sci USA 107: 3522–7.
- Moitra K, Lou H, Dean M (2011) Multidrug efflux pumps and cancer stem cells : in sight into multidrug resistance and therapeutic development. Clin Pharmacol Ther 89: 491–502.
- Frame FM, Maitland NJ (2011) Cancer stem cells, models of study and implications of therapy resistance mechanisms. Adv Exp Med Biol 720: 105–118. [crossref]
- Liau MC (2007) Pharmaceutical composition inducing cancer cell differentiation and the use for treatment and prevention of cancer thereof. US Patent 7232578 B2, June 19.
- Liau MC, Ashraf AQ, Lee SS, Burzynski SR (1990) Riboflavin as a minor active anticancer component of Antineoplaston A2 and A5. Intl J Tiss React 12: 19–26.
- Liau MC, Huang LJ, Lee JH, Chen SC (1998) Development of differentiation helper inducers for the differentiation therapy of cancer. Chin Pharm J 50: 289–303.
- Liau MC, Liau CP (2002) Methyltransferase inhibitors as excellent differentiation helper inducers for differentiation therapy of cancer. Bull Chin Cancer 166–168.
- Laerum OD, Paukovits WR (1984) Modulation of murine hemopoiesis in vivo by a synthetic hemoregulatory pentapeptide (HP5b). Differertiation 27: 106–112.
- Bourgeade MF, Besançon F (1984) Induction of 2’,5’-oligoadenylate synthetase by retinoic acid in two transformed human cell lines. Cancer Res 44: 5355–5360. [crossref]
- Das BC, Thapa P, Karki R, Das S, Mahapatra S, et al. (2014) Retinoic acid signaling pathways in development and diseases. Bioorg Med Chem 22: 673–683. [crossref]
- Cunningham TJ, Duester G (2015) Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat Rev Mol Cell Biol 16: 110–123. [crossref]
- Gudas LJ, Wagner JA (2011) Retinoids regulate stem cell differentiation. J Cell Physiol 226: 322–330. [crossref]
- Stockhausen MT, Kristoffersen K, Stobbe L, Poulsen HS (2014) Differentiation of glioblastoma multiforme stem-like cells leads to down-regulation of EGFR and EGFRviii and decreased tumorigenic and stem-like cell potential. Cancer Biol Ther 15: 216–224.
- Sulaiman G, Cooke A, French B (2017) MyD88 is an essential component of retinoic acid-induced differentiation in human pluripotent embryonal carcinoma cells. Cell Death Diff 24: 1975–1986.
- El-Houjeiri L, Saad W, Hayar B, Aouad P (2017) Antitumor effect of the atypical retinoid ST1926 in acute myeloid leukemia and nanoparticle formation prolongs lifespan and reduce tumor burden of xenograft mice. Mol Cancer Ther 16: 2047–2057.
- Heynen GJ, Nevedomskaya E, Palit S, Jagalur Basheer N, Lieftink C, et al. (2016) Mastermind-Like 3 Controls Proliferation and Differentiation in Neuroblastoma. Mol Cancer Res 14: 411–422. [crossref]
- Yang Q, Sakurai T, Kakudo K (2002) Retinoid, retinoic acid receptor beta and breast cancer. Breast Cancer Res Treat 76: 167–173. [crossref]
- Zhao X, Graves C, Ames SJ, Fisher DE, Spanjaard RA (2009) Mechanism of regulation and suppression of melanoma invasiveness by novel retinoic acid receptor-gamma target gene carbohydrate sulfotransferase 10. Cancer Res 69: 5218–5225. [crossref]
- Lotem J, Sachs L (1979) Regulation of normal differentiation in mouse and human myeloid leukemia cells by phorbol esters and the mechanism of tumor promotion. Proc Natl Acad Sci USA 76: 5158–5162.
- Cooper RA, Braunwald AD, Kuo AL (1982) Phorbol ester induction of leukemic cell differentiation is a membrane-mediated process. Proc Natl Acad Sci U S A 79: 2865–2869. [crossref]
- Weeks CE, Slaga TJ, Hemmings H, Gleason GL (1979) Inhibition of phorbol ester-induced tumor promotion in mice by vitamin A analogs and anti-inflammatory steroid. J Natl Cancer Inst 63: 401–406.
- Han ZT, Zhu XX, Yang RY, Sun JZ (1998) Effect if intravenous infusion of 12-O-tetradecanoyl phorbol-13-acetate (TPA) in patients with monocytic leukemia: preliminary studies on therapeutic efficacy and toxicity. Proc Natl Acad Sci USA 95: 5357–5361.
- Schaar DG, Goodell L, Aisner J, Cui XX (2006) Phase I clinical trial of 12-O-tetradecanoylphorbol-13-acetate for patients with relapsed/refractory malignancies. Cancer Chemother Pharmacol 57: 789–795.
- Bond JA, Gescher A, Verschoyle RD, Lemoine NR (2007) Cytotoxic action of phorbol esters on human pancreatic cancer cells. Int J Cancer 121: 1445–1454.
- Xu L, Zhang L, Hu C, Liang S (2016) WNT pathway inhibitor pyrvinium pamoate inhibits the self-renewal and metastasis of breast cancer stem cells. Intl J Oncol 48: 1175–1186.
- Stoddart A, Wang J, Hu C, Fernald AA, Davis EM, et al. (2017) Inhibition of WNT signaling in the bone marrow niche prevents the development of MDS in the Apcdel/+ MDS mouse model. Blood 129: 2959–2970. [crossref]
- Momtazi-Borojeni AA, Abdollahi E, Ghasemi F, et al. (2018) The novel role of pyrvinium in cancer therapy. J Cell Physiol 233: 2871–2881. [crossref]
- Barbarino M, Cesari D, Intruglio R, Indovina P (2018) Possible repurposing of pyrvinium pamoate for the treatment of mesothelioma: a preclinical assessment. J Cell Physiol 233: 7391–7401.
- Yao CJ, Lai GM, Chen CF (2007) Elimination of cancer stem-like “side population” cells existed in MCF-7 breast cancer cells by human urine extract CDA-2. Literature documentation on CDA-2 issued by Everlife Pharmaceutical Co, Hefei, Anhui, China, 2007; 5–11.
- Ren J (2009) Cancer stem cell: the new target for fighting cancer from the latest transitional research. Biomed Pharmaco Therapy 63: 626–630.
- Mougeri-Sacca M, Dimarino S, De Maria R (2013) Biological and clinical implication of cancer stem cells in primary brain tumors. Front Oncol 3: 6.
- Lathia JD, Venere M, Rao MS, Rich JN (2011) Seeing is believing: are cancer stem cells the Loch Ness monster of tumor biology? Stem Cell Rev 7: 227–237. [crossref]
- Fruehauf JP, Liau MC (2011) Therapeutic targeting of HIF to reverse the cancer stem cell phenotype and epithelial-mesenchymal transition. Therapy 8: 737–740.
- Caramels J, Papadogeorgakis E, Hill L, Browne GJ, Richard G, et al. (2013) A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell 24: 466–480. [crossref]
- Smigiel JM, Parameswaran N, Jackson MW (2017) Potent EMT and CSC Phenotypes Are Induced By Oncostatin-M in Pancreatic Cancer. Mol Cancer Res 15: 478–488. [crossref]
- Nomura A, Gupta VK, Daner P, Sharma NS (2018) NFkB-mediated invasiveness in CD133+ pancreatic TICs is regulated by autocrine and paracrine activation of IL1 signaling. Mol Cancer Res 16: 162–72.
- Kampalath BN, Liau MC, Burzynski B, Burzynski SR (1987) Chemoprevention by antineoplaston A10 of benzo(a)pyrene-induced pulmonary neoplasia. Drugs Exp Clin Res 1: 51–55. [crossref]
- Kampalath BN, Liau MC, Burzynski B, Burzynski SR (1990) Protective effect of Antineoplaston A10 in hepatocarcinogenesis induced by aflatoxin B1. Intl J Tiss React 12: 43–50.
- Liau MC, Chang CF, Giovanella BC (1980) Demonstration of an altered S-adenosylmethionine synthetase in human malignant tumors xenografted into athymic nude mice. J Natl Cancer Inst 64: 1071–1075.
- Knudson AG Jr (1971) Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A 68: 820–823. [crossref]
- Liau MC, Chang CF, Becker FF (1979) Alteration of S-adenosylmethionine synthetase during chemical hepatocsarcinogensis and in resulting carcinoma. Cancer Res 39: 2103–2109.
- Morley JE (2001) Hormone, aging, and endocrines in the elderly. In: Felig P, Frohman LA, “endocrinology and Metabolism, 4th (edn), McGrow-Hill, Inc., Medical Publishing Division, New York, Pg No: 1455–1482.
- Liau MC, Leong Y, Liau CP, Burzynski SR (1992) Prevention of Drug-induced DNA hypermethylation by antineoplaston components. Intl J Exptl Clin Chemother 5: 19–27.
- Bagheri V, Memar B, Behzadi R, Aliakbarian M (2018) Isolation and identification of chemotherapy-enriched sphere forming cells from a patient with gastric cancer. J Cell Physiol 233: 7036–7046.