Abstract
Kinases represent one of the most prominent classes of drug targets in current medicinal chemistry research. They are frequently over expressed in various diseases such as cancer, inflammation, or autoimmune disorders, playing critical roles in their pathophysiology. Since the early 1980s, numerous potent kinase inhibitors have been developed and approved. However, kinase inhibitors often face challenges including drug resistance and off-target effects. To overcome these limitations, novel strategies are required. Since 2013, several research groups have proposed converting potent kinase inhibitors into PROTAC molecules, leveraging cellular machinery to degrade target proteins. Results demonstrate that PROTACs significantly enhance biological effects compared to their parent inhibitors. This review focuses on recent advances in PROTAC technology applied to receptor tyrosine kinases (RTKs), with particular emphasis on compounds reported since 2018.
Keywords
Receptor tyrosine kinase; Targeted protein degradation; PROTAC; Review
Kinases are macromolecules involved in signal transduction and pathway regulation within biological systems, primarily functioning through two mechanisms: (1) Catalytic function: transferring phosphate groups from high-energy donor molecules (e.g., ATP) to specific substrates; (2) Non-catalytic functions: such as scaffold roles and allosteric regulation mediated by protein interactions. Protein kinases mainly include receptor tyrosine kinases (RTKs), non-receptor tyrosine kinases (NRTKs), and serine-threonine kinases (STKs), among other families. Dysregulated kinase activity contributes to diseases such as acute myeloid leukemia [1], melanoma [2], breast cancer [3], and prostate cancer, making them hot targets for small-molecule drug discovery. Although existing small-molecule inhibitors [4] have shown significant efficacy in clinical treatments, they possess limitations including lack of selectivity toward highly homologous kinases and the emergence of drug resistance. The rapidly developing technology of Proteolysis-Targeting Chimeras (PROTACs) is considered a promising strategy to overcome these challenges [5]. PROTACs are bifunctional molecules that induce the formation of a complex between the target protein (protein of interest, POI; here, a kinase) and an E3 ubiquitin ligase (UL). This complex mediates polyubiquitination of the POI, leading to its recognition and degradation by the ubiquitin-proteasome system (UPS) (Figure 1). Since 2013, reports on kinase-targeting PROTAC molecules have grown exponentially. This article reviews recent progress in PROTACs targeting protein kinases, which may facilitate the development of novel therapies to overcome drug resistance in cancer treatment.
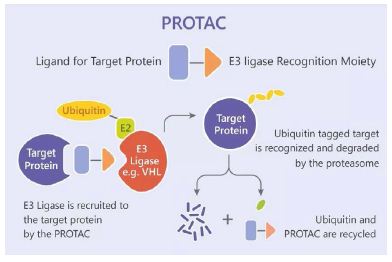
Figure 1: PROTACs induce protein ubiquitination and degradation.
Receptor tyrosine kinases (RTKs) are cell surface receptors with a similar molecular structure, comprising an extracellular ligand-binding domain, a transmembrane helix, and an intracellular region containing a tyrosine kinase domain. Genetic mutations can alter RTK activity, expression levels, cellular distribution, and regulation, potentially leading to cancers, diabetes, inflammation, and severe bone or vascular diseases. Recently, FMS-like tyrosine kinase 3 (FLT3) [6], tropomyosin receptor kinase C (TrkC) [7], anaplastic lymphoma kinase (ALK) [8,9], and epidermal growth factor receptor (EGFR) [10] have been reported to be degraded by their corresponding PROTAC molecules. Their structures are shown in Figure 2, and biological activities are summarized in Table 1.
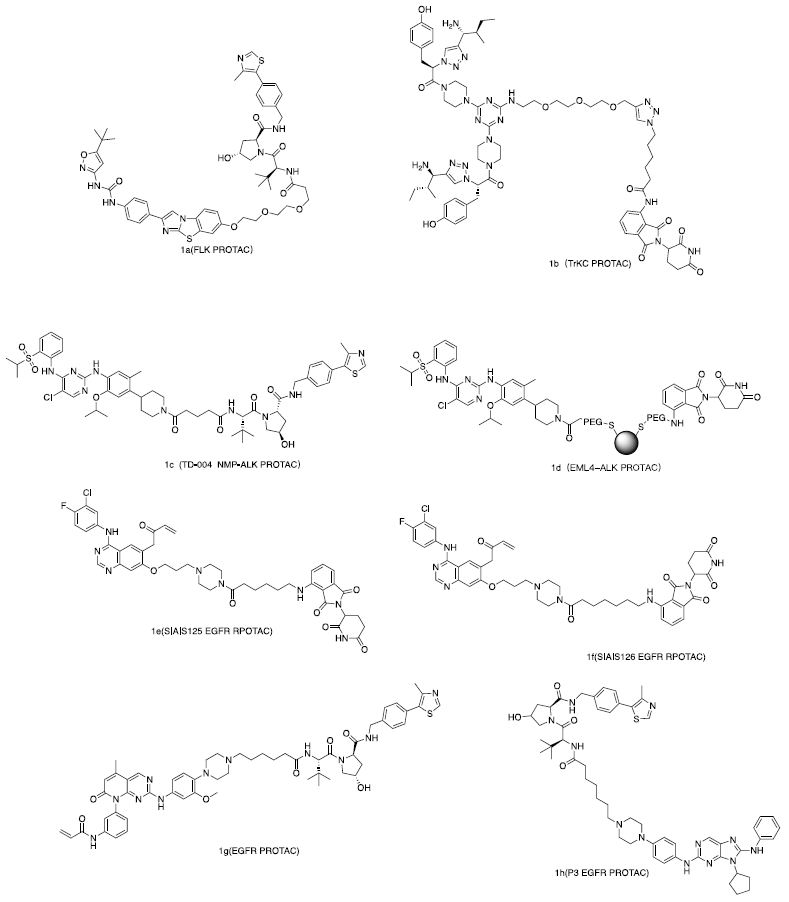
Figure 2: Reported RTKs degrader.
Table 1: Reported RTKs degrader.
|
Compounds |
Target | POI ligand | E3 Ligand |
Cells (DC50) |
| 1a | FLT3/ITD | AC220 | VHL Ligand | MOLM-14 MV4-11[6] |
| 1b | TrKC | IY-IY | Pomalidomide | Hs578t (0.1-1.0 μM) [7] |
| 1c (TD-004) | NMP-ALK | Ceritinib | VHL Ligand | SU-DHL-1 H3122 [8] |
| 1d | EML4-ALK | Ceritinib | Pomalidomide | NCI-H2228 [9] |
| 1e | EGFR | SIAIS092 | Pomalidomide | PC9(100 nM)
H1975(30-50 nM) [30] |
| 1f | EGFR | SIAIS092 | Pomalidomide | PC9(30-100 nM)
H1975(<30 nM) [30] |
| 1g | EGFR | XTF-262 | VHL Ligand | H1975(5.9±2.1 nM) [28] |
| 1h | EGFR | Ribociclib | VHL Ligand | HCC827(0.51 nM)
H1975(126.6 nM) [29] |
PROTACs Degrading FLT3
FLT3 is expressed in hematopoietic stem cells and plays a crucial role in normal hematopoiesis; its dysfunction often leads to blood disorders. Acute myeloid leukemia (AML) is a common malignant tumor of the hematopoietic system, with FLT3 gene mutations accounting for 30% of AML cases, among which internal tandem duplication (ITD) mutations are the most common. Patients carrying FLT3-ITD mutations exhibit elevated white blood cell levels and poor prognosis [1,11-13]. Therefore, FLT3/ITD is a promising target for AML treatment, and maximal sustained inhibition of this signaling is crucial for clinical response [11]. Based on this background, numerous PROTACs targeting FLT3 for degradation have emerged. The Burslem group reported a PROTAC 1a [6,11] composed of AC220 [14,15] as the warhead, VHL as the E3 ligase ligand, and connected by a PEG linker of appropriate length. This PROTAC exhibited similar cellular inhibitory activity and selectivity to AC220 in vitro but showed 3.5-fold greater inhibition of cell proliferation in MOLM-14 and MV4-11 cell lines and induced better apoptosis in leukemia cells at low doses. 1a induced FLT3-ITD degradation in vivo; in MV4-11 xenograft models, administration at 30 mg·kg⁻¹ every 24 hours for three days reduced FLT3 levels in tumors by 60%. These results suggest that degradation of FLT3-ITD may provide a useful therapeutic approach for AML.
PROTACs Degrading TrkC
Trk kinases are a family of protein tyrosine kinases with high affinity for neurotrophin (NT) growth factors, which are essential for neuronal differentiation and survival in the peripheral and central nervous systems. TrkC is overexpressed in many human tumors, particularly in neuroblastoma [12], glioblastoma [12], breast cancer [16], and melanoma [2]. Its aberrant activation promotes cell growth and metastasis in certain forms of tumorigenesis [3], making effective reduction of TrkC expression significant for treating these cancers. In 2019, Zhao and Burgess reported the first PROTAC 1b degrading TrkC [7]. This PROTAC uses a bivalent peptide (isoleucine-tyrosine-tyrosine-isoleucine) analog as the warhead, which binds TrkC with submicromolar affinity and facilitates good cellular internalization [17,18]. The warhead is connected to pomalidomide via a PEG linker. 1b degraded TrkC in Hs578t cells at concentrations of 1-10 μM, with a DC₅₀ of 0.1-1.0 μM [7]. This PROTAC demonstrates the applicability of the technology to reduce TrkC levels and provides insights for developing more PROTACs, such as those using FDA-approved inhibitors like larotrectinib or entrectinib as warheads [19,20], potentially yielding highly effective molecules.
PROTACs Degrading ALK
ALK is part of the insulin receptor family. Although its exact function in mammalian cells is not fully understood, various forms of ALK fusion proteins are known to drive oncogenesis in multiple cancers. For example, the NMP-ALK fusion is commonly found in anaplastic large cell lymphoma (ALCL) [21,22]. Numerous studies indicate that inhibiting ALK activity suppresses the proliferation of cancer cells driven by ALK fusions [23,24]. Thus, developing ALK-targeted PROTACs appears to be a promising approach to enhance the efficacy of approved ALK inhibitors. The Kang group reported a PROTAC TD-004 [8] using ceritinib as the warhead [25] and connected to a VHL ligand via a long linker containing an isopropyl chain and an amide bond. TD-004 degraded approximately 90% of NMP-ALK fusion protein in SU-DHL-1 cells, with IC₅₀ values of 58 nM and 180 nM in ALK-positive SU-DHL-1 and H3122 cell lines, respectively, while showing an IC₅₀ > 1000 nM in ALK-low A549 cells, indicating significant selectivity [8]. In H3122 xenograft models, daily administration of 58 mg·kg⁻¹ for 14 days significantly reduced tumor volume without causing notable weight loss. Besides TD-004, Zhang et al. [26] and Powell et al. [27] reported other ceritinib-based PROTACs using pomalidomide as the E3 ligase ligand. These PROTACs differed mainly in linker length and composition, exhibiting stronger anti-proliferative activity in vitro than TD-004 but lacking in vivo validation. These examples illustrate that even with the same warhead, variations in linker length, composition, and E3 ligase ligand can significantly impact PROTAC bioactivity. Besides NMP-ALK, another ALK fusion protein, EML4-ALK, was targeted by a degrader reported by the Liu group. This multivalent PROTAC 2d consists of multiple warheads and E3 ligase ligands attached to gold nanoparticles [9]. Due to the strong affinity of gold nanoparticles for thiols, modified ceritinib and pomalidomide were easily conjugated to the nanoparticle surface to form multivalent PROTACs. Cellular analysis showed that incubation with NCI-H2228 cells for 24 hours reduced EML4-ALK levels by 80%, with prominent anti-proliferative activity (IC₅₀ = 4.8 μM), while negligible cytotoxicity was observed in ALK-negative A549 cells. Although in vivo efficacy requires further validation, the multivalency offered by gold nanoparticles provides an effective strategy to bring warheads and E3 ubiquitin ligases into proximity, facilitating ternary complex formation.
PROTACs Degrading EGFR
Activating mutations in EGFR are closely associated with non-small cell lung cancer (NSCLC). However, even with FDA-approved third-generation EGFR-TKIs like osimertinib, resistance eventually develops, reducing therapeutic efficacy. In April 2020, Zhang et al. [28] reported a series of PROTACs selectively targeting mutant EGFR. One of the most potent compounds, 1g, selectively degraded EGFRL858R/T790M with a DC₅₀ of 5.9 nM while sparing the wild-type protein. In December of the same year, Zhao et al. [29] reported a series of VHL-based PROTACs, among which compound P3 showed potent anti-proliferative activity in HCC827 and H1975 cells with IC₅₀ values of 0.83 nM and 203.01 nM, respectively, and DC₅₀ values of 0.51 nM and 126.2 nM for EGFRdel19 and EGFRL858R/T790M, respectively. Besides inhibiting EGFR signaling, P3 significantly induced apoptosis, arrested the cell cycle, and inhibited colony formation. In June 2021, Qu et al. [30] reported SIAIS125 and SIAIS126—two PROTACs composed of the EGFR inhibitor canertinib and pomalidomide connected by linkers of different lengths. These degraders selectively degraded EGFRL858R/T790M in H1975 cells and EGFREx19del in PC9 cells for up to 72 hours, also inducing significant apoptosis, cell cycle arrest, and growth inhibition. They did not degrade the EGFREc19del/T790M mutant in PC9BRca1 cells or wild-type EGFR in A549 lung cancer cells. Since the first report of kinase-targeted PROTACs in 2013, many research groups have proposed converting potent kinase inhibitors into PROTACs. This technology, which harnesses cellular machinery to degrade proteins, has propelled several compounds to the forefront of drug development, offering advantages for enhancing therapeutic efficacy. PROTACs may yield superior biological outcomes compared to parent inhibitors; for instance, FLT3-PROTACs induce apoptosis more effectively in leukemia cells, and BCR-ABL degraders exhibit longer-lasting inhibition of BCR-ABL and its downstream signaling than dasatinib. PROTACs may achieve higher selectivity than ATP-competitive kinase inhibitors, as seen with CST620 selectively targeting CDK6. They can degrade proteins that have developed resistance to inhibitors due to mutations, such as the aforementioned EGFR degraders. PROTACs can utilize allosteric inhibitors as POI recruiters to enhance selectivity and reduce side effects of parent inhibitors; for example, GMB-475, using GNF-2 as the warhead, degrades the target kinase while abolishing its non-kinase functions (scaffold roles), deepening our understanding of the protein’s role in signaling networks. PROTACs can degrade membrane-bound proteins associated with various diseases, such as JAK degraders JP-1 and JP-2. PROTACs synthesized using reversible covalent inhibitors as POI recruiters may retain the reversible covalent binding characteristics of the parent inhibitors, slowing displacement by competitors. Rapid synthesis methods for PROTACs have been reported, reducing the time cost of degrader synthesis and facilitating the design of degraders for other targets.
Compared to small molecules, PROTACs offer numerous advantages. However, developing in vivo effective PROTACs remains a challenge for medicinal chemists. Although PROTACs can be viewed as combinations of POI ligands, linkers, and E3 ubiquitin ligase ligands, the aforementioned reports confirm that random combinations do not yield预设 effects. In the design and synthesis of PROTACs, the choice and structural modification of the POI ligand, the chemical composition and length of the linker, and the selection of the E3 ligase ligand can significantly impact their efficacy. Therefore, in-depth structure-activity relationship studies are necessary to discover the most active structures, which may require considerable time. Another critical challenge is the in vivo evaluation of PROTACs. Their large molecular weight places them outside the realm of traditional small molecules, and the flexibility and chemical composition of the linker make them susceptible to in vivo environmental interference, leading to poor stability, as seen with MT802. However, optimization has yielded PROTACs with improved pharmacokinetic parameters and orally available degraders, addressing stability issues to a great extent. In 2019, the first PROTACs entered clinical trials, and several kinase-targeted candidates are expected to follow soon. To date, the use of thalidomide and its analogs as E3 ligase ligands remains the most common approach. However, these agents can lead to degradation of lymphoid transcription factors like IKZF1 and IKZF3, potentially affecting the hematopoietic system. Moreover, cereblon (CRBN) is not essential in most cancer cell lines, and mutations in this ligase could confer resistance to PROTACs. Thus, the discovery of novel E3 ligase ligands is extremely important. In summary, PROTAC technology is a suitable tool for generating active compounds for treating various diseases. Currently, most reported PROTACs are based on well-established positive compounds and commonly used FDA-approved drugs, somewhat limiting the technology’s full potential. This strategy will likely be applied in the coming years to target currently undruggable targets, playing a crucial role in revealing new clinical targets and providing treatments for many diseases. Therefore, although this rapidly growing research field is still in its early stages, it offers encouraging directions for both biological understanding and the future of medicinal chemistry.
References
- Birg F, Courcoul M, Rosnet O, Bardin F, Pébusque MJ, et al. (1992) Expression of the FMS/KIT-like gene FLT3 in human acute leukemias of the myeloid and lymphoid lineages. Blood 80: 2584-2593. [crossref]
- Xu X, Tahan SR, Pasha TL, Paul JZ (2003) Expression of neurotrophin receptor Trk-C in nevi and melanomas. J Cutan Pathol 30: 318-22. [crossref]
- Jin W, Kim GM, Kim MS, Mi HL, Chohee Y, et al. (2010) TrkC plays an essential role in breast tumor growth and metastasis. Carcinogenesis 31: 1939-1947. [crossref]
- Bhullar K. S., Lagaron N. O., Mcgowan E. M., Indu Parmar, Amitabh J, et al. (2018) Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer 17. [crossref]
- Groppe JC (2019) Induced degradation of protein kinases by bifunctional small molecules: a next-generation strategy. Expert Opin Drug Discov 14: 1237-1253. [crossref]
- Burslem GM, Song J, Chen X, John H, Craig MC (2018) Enhancing Antiproliferative Activity and Selectivity of a FLT-3 Inhibitor by Proteolysis Targeting Chimera Conversion. J Am Chem Soc, 140: 16428-16432. [crossref]
- Zhao B, Burgess K (2019) TrkC-Targeted Kinase Inhibitors And PROTACs. Mol Pharm 16: 4313-4318.
- Kang CH, Lee DH, Lee CO, Jae DH, Chi HP, et al. (2018) Induced protein degradation of anaplastic lymphoma kinase (ALK) by proteolysis targeting chimera (PROTAC). Biochem Biophys Res Commun 505: 542-547. [crossref]
- Wang Y, Han L, Liu F, Fubai Y, Xueyang J et al. (2020) Targeted degradation of anaplastic lymphoma kinase by gold nanoparticle-based multi-headed proteolysis targeting chimeras. Colloids Surf B Biointerfaces 188. [crossref]
- Sigismund Sara, Avanzato Daniele, Lanzetti Letizia (2018) Emerging functions of the EGFR in cancer. Mol Oncol 12: 3-20. [crossref]
- Pratz K. W., Cortes J., Roboz G. J., et al. (2009) A pharmacodynamic study of the FLT3 inhibitor KW-2449 yields insight into the basis for clinical response. Blood 113: 3938-3946. [crossref]
- Wang Y, Hagel C, Hamel W, Niranjan R, Omotayo, A et al. (1998) Trk A, B, and C are commonly expressed in human astrocytes and astrocytic gliomas but not by human oligodendrocytes and oligodendroglioma. Acta Neuropathol 96: 357-364.
- Jiang Wei, Gao Yujuan, Su Yanhua (2020) Research progress on FLT3 gene mutation in acute myeloid leukemia. Journal of Clinical and Pathological Medicine 403:718-722.
- Zarrinkar PP, Gunawardane RN, Cramer MD, Michael FG, Daniel B, et al. (2009) AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood 114: 2984-2892. [crossref]
- Chao Q, Sprankle KG, Grotzfeld RM, Andiliy GL, Todd AC, et al. (2009) Identification of N-(5-tert-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1 ,3]benzothiazol-2-yl]phenyl}urea dihydrochloride (AC220), a uniquely potent, selective, and efficacious FMS-like tyrosine kinase-3 (FLT3) inhibitor. J Med Chem, , 52: 7808-78016. [crossref]
- Blasco-Gutierrez MJ, Jose-Crespo IJ, Zozaya-Alvarez E, Rafael RS, Natividad GA (2007) TrkC: a new predictive marker in breast cancer?. Cancer Invest 25: 405-410. [crossref]
- Chen D, Brahimi F, Angell Y, Yu-CL, Jennifer M, et al. (2009) Bivalent peptidomimetic ligands of TrkC are biased agonists and selectively induce neuritogenesis or potentiate neurotrophin-3 trophic signals. ACS Chem Biol 4: 769-781. [crossref]
- Chen J, Wang X, He F, Zhengying P (2018) Development of a Selective Labeling Probe for Bruton’s Tyrosine Kinase Quantification in Live Cells. Bioconjug Chem 29: 1640-1645. [crossref]
- Drilon A, Nagasubramanian R, Blake JF, Nora K, Brian BT, et al. (2017) A Next-Generation TRK Kinase Inhibitor Overcomes Acquired Resistance to Prior TRK Kinase Inhibition in Patients with TRK Fusion-Positive Solid Tumors. Cancer Discov 7: 963-72. [crossref]
- Siena S, Drilon A, Ou I, et al. (2015) 29LBA Entrectinib (RXDX-101), an oral pan-Trk, ROS1, and ALK inhibitor in patients with advanced solid tumors harboring gene rearrangements. Eur J Cancer 51.
- Hallberg B, Palmer RH (2013) Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer 13: 685-700. [crossref]
- Pulford K, Lamant L, Morris SW, Butler LH, Wood KM, et al. (1997) Detection of anaplastic lymphoma kinase (ALK) and nucleolar protein nucleophosmin (NPM)-ALK proteins in normal and neoplastic cells with the monoclonal antibody ALK1. Blood 89: 1394-1404. [crossref]
- Christensen JG, Zou HY, Arango ME, Qiuhua L, Joseph HL et al. (2007) Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma . Mol Cancer Ther 6: 3314-3322. [crossref]
- Soda M, Choi YL, Enomoto M, Shuji T, Yoshihiro Y, et al. (2007) Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448: 561-566. [crossref]
- Roskoski R Jr (2020) Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update. Pharmacol Res 152.
- Zhang C, Han XR, Yang X, Biao J, Jing L, et al. (2018) Proteolysis Targeting Chimeras (PROTACs) of Anaplastic Lymphoma Kinase (ALK). Eur J Med Chem 151: 304-14. [crossref]
- Powell CE, Gao Y, Tan L, Katherine AD, Radosław PN, et al. (2018) Chemically Induced Degradation of Anaplastic Lymphoma Kinase (ALK). J Med Chem 61: 4249-4255. [crossref]
- Zhang X, Xu F, Tong L, Tao Z, Hua X, et al. (2020) Design and synthesis of selective degraders of EGFR(L858R/T790M) mutant. Eur J Med Chem 192. [crossref]
- Zhao HY, Yang XY, Lei H, Xiao-XX, She ML, et al. (2020) Discovery of potent small molecule PROTACs targeting mutant EGFR. Eur J Med Chem 208. [crossref]
- Qu X, Liu H, Song X, Ning S, Hui Z, et al. (2021) Effective degradation of EGFR(L858R+T790M) mutant proteins by CRBN-based PROTACs through both proteosome and autophagy/lysosome degradation systems. Eur J Med Chem 218. [crossref]