DOI: 10.31038/JMG.2020315
Abstract
Coronary Artery Disease (CAD) is the number one cause of death in the world. Epidemiologists’ claim 50 percent of CAD is genetic. The identified first genetic risk variant, 9p21, was discovered in 2007, and subsequently, efforts have led to identifying hundreds of genetic risk variants predisposing to CAD. CAD is preventable based on clinical trials showing reduction in conventional risk factors, such as cholesterol, is associated with 30–40 percent reduction in cardiac mortality and events. Statin therapy, which lowers plasma cholesterol, is very safe and effective. About 50 percent of all Americans living a normal lifespan will experience a cardiac event. The challenge is selecting among asymptomatic individuals, the 50 percent who would benefit most from prevention. Conventional risk factors are age-dependent, while genetic risk variants are independent of age, and can be determined anytime from birth on, since one’s DNA does not change in one’s lifetime. Utilizing a microarray containing the genetic risk variants, and DNA from saliva or blood, studies was performed in over 1 million cases and controls. Genetic risk variants were shown to be relatively independent of conventional risk factors and offer greater discriminatory power in stratifying for CAD risk. Individuals with the highest genetic risk score (GRS) had the highest risk for CAD and benefitted most from statin therapy. A recent study employed the genetic risk in a sample size of 55,685 individuals. Those with a high GRS for CAD (20%) had a 91 percent higher risk for cardiac events. Individuals with a healthy lifestyle and high GRS had a 46 percent lower risk for cardiac events in comparison to those with an unfavorable lifestyle thus, genetic risk can be reduced. Utilizing the GRS to risk stratify for primary prevention of CAD will represent a paradigm shift in halting the spread of this pandemic disease.
Keywords
genetics, coronary artery disease, risk prediction of CAD, prevention of CAD
Comprehensive Risk (Genetic and Acquired) Stratification for Primary Prevention of CAD
Introduction
Coronary artery disease (CAD) is now well established as the number one cause of death in the world, accounting for about 1/3 of all deaths, equally affecting males and females. While, CAD was, until recently, primarily a disease of the western world, it is now pandemic involving low, middle, and high income countries [1]. While, the disease has been decreasing in the west, there has been a marked increase in the east. It is estimated that about 50% of males or females, who live a normal lifespan in the U.S, will be expected to encounter at least one cardiac event [1]. Similar statistics apply to most of the western world. Secondary (already have had a cardiac event) prevention has been very successful by modifying the conventional risk factors predisposing to CAD (cholesterol, hypertension, diabetes, smoking, sedentary lifestyle, obesity, and family history). These studies confirm that CAD is preventable, however to halt the pandemic spread one will have to initiate primary prevention. A major challenge is to identify among asymptomatic individuals those who will benefit most from preventive therapy for CAD. The recent discovery of genetic risk variants predisposing to CAD offers a new tool to select and motivate asymptomatic individuals at risk for CAD.
The Pursuit of Genetic Variant Predisposing to CAD
Epidemiologists have for decades, claimed that approximately 50 percent of predisposition for CAD is inherited [2–4]. Most common chronic diseases are claimed to have similar genetic predisposition. CAD is a polygenic disorder in which genetic predisposition is transmitted by multiple genes, each with only minimal risk. The human genome has 3.2 billion base pairs, referred to as nucleotides. Sequencing studies have shown that, comparison of any two human genomes show a variation in DNA sequence of only 1 percent, with 99 percent being identical [5–8]. Most of this 1 percent difference is due to large structural variants, such as inversions, or copy number variations, with the remainder being due to single nucleotide polymorphisms (SNP). The number of SNPs in the genome is fairly constant, at approximately 5 million per genome [9]. While, the SNPs account for less than 1/10th of 1 percent of the human genome sequences, they are claimed to be responsible for 70–80 percent [9] of the variation underlying the unique features of human beings (eg color of your hair, predisposition to disease).
To pursue the genetic variants predisposing to CAD, it was necessary to adopt an unbiased approach which would require DNA markers, spanning the whole human genome. The most desirable would be, having markers evenly distributed throughout the genome, at intervals of 3,000 to 6,000 bases, which would require at least one million DNA markers. The millions of SNPs annotated by HapMap [10,11] provided the necessary DNA markers to span the genome. Secondly, in designing the study we adopted the Case Control Association Approach, in which the frequency of markers in the cases would be compared to that of controls [12–13]. This would be referred to as a Genome Wide Association Study (GWAS). Any DNA marker that occurred more frequently in the cases, than in controls, would indicate it is a risk for CAD, or is in close physical proximity to a sequence that increases risk for CAD. This approach of using a million markers would present a major problem if we accepted a p-value of 0.05, in comparing the frequency of a marker in cases versus controls, as it would result in 50,000 false positives. Thus, a statistical correction was necessary. We agreed that the most stringent would be a Bonferroni whereby the p-value of 0.05 would be divided by one million, giving a required p-value of 5×10–8. This p-value subsequently became known as genome wide significant and further enhanced the need for a large sample size. A further requirement was all SNPs associated with increased risk for CAD of genome wide significance had to be confirmed by replication in an independent population.
Discovery of 9p21 as a Risk Variant for CAD and the Formation of an International Consortium
In our initial attempt to identify the first genetic risk variant predisposing to CAD we hypothesized that CAD as a common disorder would be due to genetic variants that occurred frequently worldwide with each variant conferring minimal risk. We adopted the Case Control Association Approach which had been shown to be successful in diabetes [14] and selected 100,000 SNPs as markers to span the genome. Also SNPs showing statistical significance required replication in one or more independent populations. The discovery of 9p21 was the result of genotyping a total population of over 23,000 individuals from Canada, US, and Denmark [15]. Simultaneously and independently, the Icelandic group also identified 9p21 [16]. Shortly thereafter, 9p21 was confirmed by the Wellcome Trust Group Case Control Consortium (WTGCCC) [17] and subsequently in many other cohorts of Europeans and East Asians [18].
The name, 9p21, refers to being on chromosome 9, the short arm, at band 2.1. 9p21 occurs in about 75 percent of individuals of European ancestry, with each copy associated, with a 25 percent increase in relative risk for CAD. It is of note, that 9p21, was found to be independent of all known risk factors for CAD, implying that risk factors, other than the known conventional risk factors, contributed to the pathogenesis of CAD. The observation that 9p21 imparted only minimal risk confirmed our hypothesis that there would be multiple risk variants, each exerting only minimal increased risk. This further enhanced our need for a large sample size. This lead to the formation of an international consortium, referred to as CARDIoGRAM [19], and subsequently with other additional investigators known as CARDIoGRAMplusC4D. The initial sample size consisted of 22,233 cases and 64,762 controls, with a replication in an independent population of 56,682 [19].
A decade later, the CARDIoGRAMplusC4D investigators, along with other independent investigators, have identified over 200 genetic risk variants predisposing to CAD, which has been summarized in several reviews [20–24]. All of these risk variants are of genome wide significance and have been replicated in an independent population. There were hundreds of other genetic risk variants which did not reach the genome wide significance (P-value of 10–8), but did satisfy the statistical criteria of false discovery of less than 5 percent. If one combines the risk variants obtained by genome wide association studies using either the corrected 10–8, or false discovery rate of 5 percent significance, they would account for 30–40 percent of the expected heritability of CAD [24].
Genetic Risk Variants for CAD are Multiple and Occur Commonly
Table 1 shows some of the features of genetic risk variants predisposing to CAD. They are very common, and over 50 percent of them occur in more than 50 percent of the population. This is in keeping with the original hypothesis, that genetic variants predisposing to common diseases would be transmitted by DNA variants that occur commonly. (2) Each genetic risk variant transmits only minimal increase in relative risk, which on the average for CAD, is less than 10 percent. (3) Over 75 percent of the genetic risk variants occur in non-protein coding regions and tend to cluster in regulatory elements. Thus, the influence of these genetic variants is mediated through regulation of protein coding sequences, upstream or downstream (cis acting), and possibly through interacting with protein coding region on other chromosomes (trans acting). Lastly, about two-thirds of the variants do not mediate their risk through any of the known conventional risk factors. Elucidation of the molecular pathways by which these unknown genetic risk variants mediate their risk, will contribute significantly to our understanding of the pathogenesis of CAD and provide targets for development of novel therapy.
Table 1. The Features of Genetic Risk Variants for CAD.
|
|
|
|
Total Genetic Risk Burden of CAD is Proportional to the Number of Risk Variants Inherited
Since, each genetic risk variant contributes only about an 8 percent increase in risk for CAD; the total genetic risk burden is proportional to the number of risk variants inherited, rather than any single variant. A single number for genetic risk of CAD can be obtained by knowing the number of genetic risk variants inherited by each individual, and taking into account the risk transmitted by each variant for CAD. The Genome-Wide Association Studies (GWAS), have previously identified the increased risk for each variant. Utilizing blood or saliva, one can genotype the extracted DNA for the number of genetic variants inherited by any one individual. The range for each single genetic risk variant is zero to two. It is zero (neither of the parent’s has the genetic variant), 1 (if only one of the parents transmits the genetic variant), and 2 (if both of the parents transmit the genetic variant). In accounting for the risk, it is standard practice to determine the weighted risk by multiplying the number of copies of each genetic risk variant times the natural log of the odds ratio [25]. The number resulting from summation of these products is the numeric Genetic Risk Score (GRS).
Clinical Trials Indicate CAD is a Preventable Disease
Several risk factors have been recognized since the 1960s that predispose to CAD. A central culprit in the pathogenesis of coronary atherosclerosis is that of cholesterol primarily due to plasma LDL (Low-Density Lipoproteins) cholesterol. Multiple trials over the past three decades have documented 30 to 40 percent decrease in cardiac events associated with decreasing plasma LDL-C [26, 27]. Other risk factors for CAD include hypertension, diabetes, smoking, age, family history, and sedentary way of life. Reduction in risk of these factors by lifestyle changes along with certain drugs has consistently shown a marked reduction in cardiac events due to CAD. Most of the studies have been performed in individuals who have already experienced a cardiac event, thus consist of secondary prevention of recurring events. Nevertheless, studies of primary prevention, primarily decreasing plasma LDL-C and controlling hypertension consistently show decreased cardiac events [28].
Evaluation of the Genetic Risk Score to Risk Stratify for CAD in the Implementation of Primary Prevention
One of the ultimate aims for pursing genetic risk for CAD was to develop a more sensitive and appropriate technique to select individuals who would benefit most from primary prevention. It is obvious that patient selection for secondary prevention simply requires proof the individuals has had a cardiac event or has significant coronary atherosclerosis. Selecting asymptomatic individuals without a cardiac history is a much greater challenge. One might champion treating everyone with a statin drug who has increased plasma LDL-C. However, the level of plasma LDL-C currently used as a target for effective control is ≤ 70 mg/dl [29–31]. This is confounded by the observation that the average plasma LDL-C in an American male in his 40s is 147 mg/dl and in a female is 121 mg/dl [29–31]. Thus, do you want to treat everyone with a statin, knowing only 50 percent will benefit? Nevertheless, Mendelian randomization studies show it is crucially important to lower the plasma LDL-C earlier in life rather than at mid-age or later in males. Ference et al. in a recent Mendelian randomization study showed a 55 percent reduction in cardiac risk for each mmol/L (38.7 mg/dl) reduction in plasma LDL-C [32]. In contrast to clinical trials in which exposure is usually for 5 years, the observed average is 20 percent reduction of risk for CAD for each mmol/L reduction in plasma LDL-C [33]. The reciprocal of this observation was also determined from longitudinal meta-analysis showing the risk for CAD doubles for each additional 10 years of exposure to increased plasma LDL-C [34].
The conventional risk factors, such as cholesterol, that we currently use for primary prevention are unfortunately age dependent and become more accurate and sensitive as one gets older. This is illustrated for the number one risk factor for CAD, namely plasma LDL-C (see table 2) The genetic risk factors if shown to be effective predictors of genetic risk for CAD would have the advantage of being independent of age, since one’s DNA does not change in a lifetime.
Table 2. Lifetime Changes in Plasma LDL-C.
|
The plasma LDL-C of neonate |
21–39 mg/dl |
|
Second decade of life |
90 mg/dl |
|
Fourth decade of life |
130–150 mg/dl |
The first study to evaluate the GRS as a means of stratifying for CAD risk was that of Mega [35]. This study performed in 2015 utilized only 27 genetic risk variants for CAD to genotype a sample size of 48,421 individuals. The study population consisted of four clinical trials, including Jupiter, which evaluated the role of decreasing plasma LDL-C, with a statin. Two of the trials involved secondary prevention and the other two involved primarily prevention. The GRS stratified the individuals into high, intermediate, and low risk. The high risk group identified by GRS was also the same group that had the most effect from statin therapy. The GRS was equally effective in identifying individuals for primary or secondary prevention. Furthermore, individuals with a high GRS required treatment of only 25 individuals with statin to prevent one cardiac event. This is in contrast to over 100 that would require treatment if stratified on the basis of known conventional risk factors. Similar results were obtained upon genotyping individuals in the West of Scotland Coronary Prevention Study (WOSCOPS). Individuals in the high genetic risk group exhibited a risk reduction of 44 percent versus a relative risk reduction of only 24 percent in others [36]. Moreover, in order to prevent one coronary event with statin therapy, those in the high genetic risk group required treatment of 13 versus 38 in the low risk group. The results confirmed that GRS has higher efficacy and is more discriminatory in risk stratification of CAD than conventional risk factors. The GRS was better at discriminating who is at a higher risk for CAD and identified individuals who would benefit the most from statins.
Risk Stratification for CAD Utilizing a Polygenic Score
The current genetic risk variants account for only about 30–40 percent of inherited risk for CAD. The inclusion of more genetic risk variants would be expected to further enhance the power of prediction and risk stratification of CAD. One technique by Inouye [37] was to include less stringent statistics, such as a false discovery rate of only 5 percent. This gave rise to a microarray containing 1.7 million genetic risk variants. The other approach by [38] included a technique that predicts association with CAD followed by further pruning to ensure exclusion of linkage disequilibrium; this microarray contains 6.6 million genetic risk variants.
Risk stratification was performed in a test set with a large sample size of 288,978 from the UK biobank utilizing the 6.6 million microarrays. Analysis showed, 8 percent of the population inherited a threefold increased risk for CAD, and 0.5 inherited a fivefold increased risk for CAD. Conventional risk factors would not have identified the 8 percent with the threefold increased risk. For example, hypercholesterolemia was present in only 20 percent of the individuals with only threefold risk in CAD, hypertension was present in only 28 percent of the group with threefold increased risk, and family history was present in only 35 percent of the threefold increased risk group.
The 1.7 microarray was used to genotype a UK biobank sample size of nearly 500,000. The top 20 percent risk group had a fourfold increased risk for CAD. These results and those of Khera et al., [38] confirm increased predictive power over that of the previous microarrays utilizing far less genetic risk variants.
Genetic Risk for CAD is Markedly Reduced by Lifestyle Changes and Drug Therapy
Multiple studies show increased discriminatory power for risk stratification of CAD utilizing microarrays containing genetic risk variants. However, this will all be for naught, if one cannot modify and reduce genetic risk. There is often the myth circulating among the populous that, if it is in your gene, you cannot do anything about it. This, of course, is a myth that has been proven by multiple therapies. Nevertheless, it is important to show that the genetic risk predicted for CAD can indeed be reduced by appropriate therapies. The first study to comprehensively assess the effect of lifestyle changes on genetic risk for CAD was published by [39]. The sample size consisted of 55,685 participants. The endpoint was favorable lifestyle, versus unfavorable lifestyle, with the former defined as no current smoking, no obesity, a healthy diet, and frequent exercise, versus a lifestyle with at least two of these bad components [39]. Risk stratification using the GRS showed, those with a high GRS for CAD (20 percent) had a 91 percent higher risk of cardiac events, than those with low GRS. Individuals with a healthy lifestyle and a high GRS had a 46 percent lower risk of cardiac events than an unfavorable lifestyle
Tikkanen et al [40] assessed the effect of physical activity on the genetic risk for CAD as determined by GRS. The sample size was that of 468,095 individuals obtained from the UK biobank. Physical activity consisted of hand grip for 3 seconds and cardiorespiratory fitness determined by oxygen consumption during cycle ergometer on a stationary bike. The higher level of physical activity was associated with less CAD in each of the low, intermediate, and high risk categories. The highest GRS had the most benefit from cardiorespiratory exercise, with 49 percent lower risk for CAD.
Genetic risk stratification for CAD has been assessed by several different investigators involving over 1 million participants and found to be highly discriminatory. Perhaps more important, genetic risk for CAD is significantly reduced by lifestyle changes and drugs. These studies refute the myth, that genetic risk cannot be attenuated or eliminated.
Genetic Risk Score, a Paradigm Shift to Halt the Epidemic of CAD
The current methods of assessing the risk for CAD by the ACA/AHA, specifically the Framingham risk score, the Randall score, and the pooled cohort equations, are not independent of other confounding factors such as age and conventional risk factors. However, GRS is completely independent of an individual’s age, based on the fact that one’s germline DNA does not change over one’s lifespan. Additionally, the GRS is relatively independent of conventional risk factors for CAD since only one third of them act through known risk mechanisms. GRS is required for a comprehensive risk assessment given that 40–60 percent of the risk of CAD is due to genetic factors. Furthermore, GRS correlates closely with sub-clinical CAD, in contrast to the lack of correlation between conventional risk factors and sub-clinical CAD [36]. Lastly, GRS is comparatively less expensive and minimally invasive; and testing can be obtained through blood or saliva samples (figure 1).
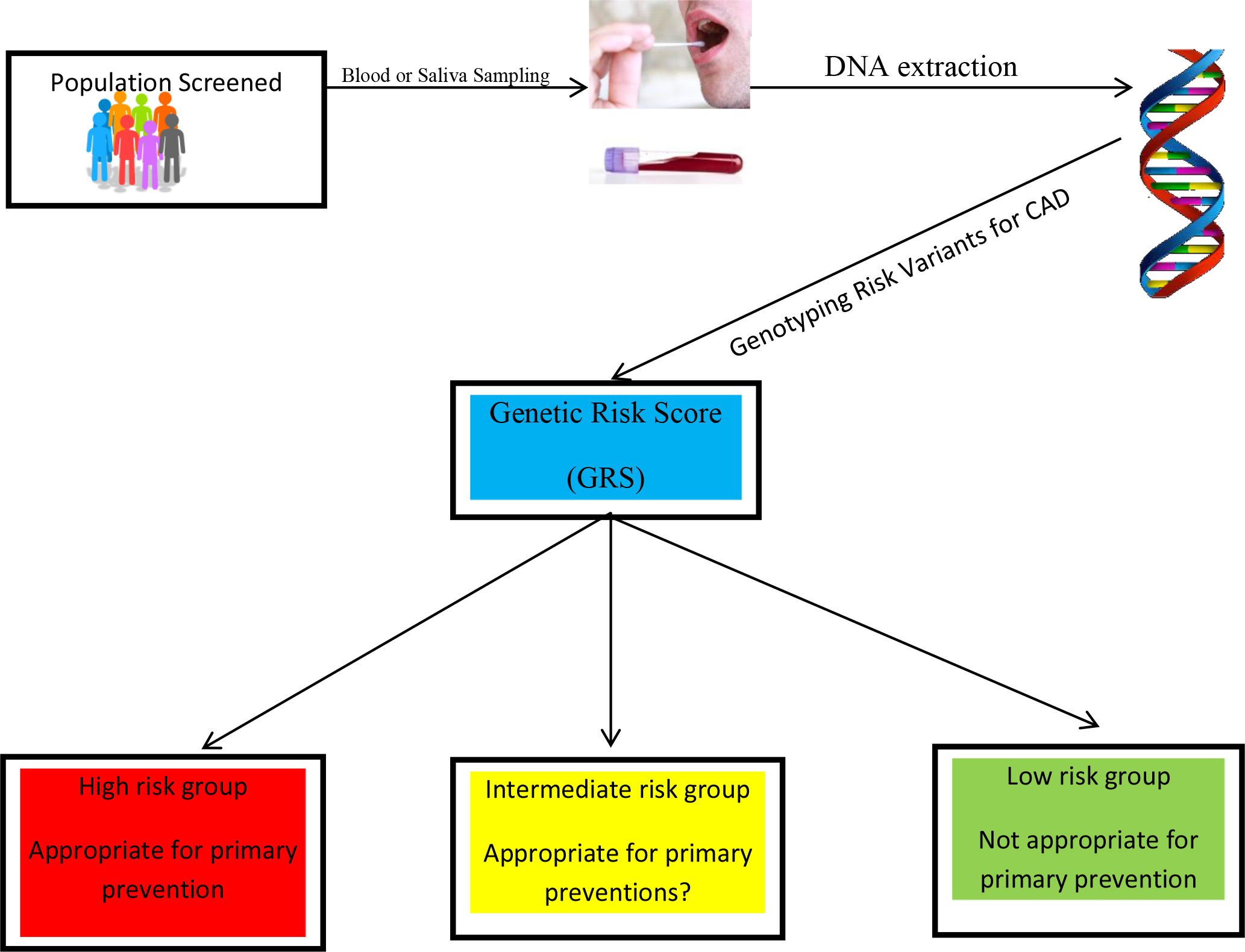
Figure 1. The sample can consist of saliva or blood. The DNA will be genotyped for the genetic risk variants predisposing to CAD. The genetic risk score (GRS) is calculated as a single number to provide the three categories high, intermediate, and low risk.
Documentation that genetic screening is effective for risk stratification of CAD is complemented by the observation that changes in lifestyles and drug therapy are extremely effective in reducing the risk. Given we have a CAD pandemic that in many countries is still increasing, beckons the need for progressive prevention based on comprehensive risk stratification using both the GRS and conventional risk factors for CAD. Risk stratification followed by primary prevention in premenopausal women has the potential to markedly attenuate the development of CAD. A similar approach adopted at an earlier age in males would be expected to have an analogous outcome.
Acknowledgment
The author has received research funding from the Canadian Institutes of Health Research, CIHR no. MOP P82810, the Canada Foundation for Innovation, the CFI, no. 11966, and the Dignity Health Foundation.
Reference
- Murray CJ, Lopez AD (2013) Measuring the global burden of disease. N Engl J Med 369: 448–457. [crossref]
- Zdravkovic S, Wienke A, Pedersen NL, Marenberg ME, Yashin AI et al. (2002) Heritability of death from coronary heart disease: a 36-year follow-up of 20966 Swedish twins. J Intern Med 252: 247–254. [crossref]
- Wienke A, Holm NV, Skytthe A, Yashin AI (2001) The heritability of mortality due to heart diseases: a correlated frailty model applied to Danish twins. Twin Res 4: 266–274. [crossref]
- Marenberg ME, Risch N, Berkman LF, Floderus B, de Faire U (1994) Genetic susceptibility to death from coronary heart disease in a study of twins. N Engl J Med 330: 1041–1046. [crossref]
- Chaisson MJP, Sanders AD, Zhao X, Malhotra A, Porubsky D et al. (2019) Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat Commun 10: 49–68. [crossref]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A et al. (2015) An integrated map of structural variation in 2,504 human genomes. Nature 526: 75–81. [crossref]
- Chaisson MJ, Wilson RK, Eichler EE (2015) Genetic variation and the de novo assembly of human genomes. Nat Rev Genet 16: 627–40. [crossref]
- Eichler EE (2019) Genetic variation, comparative genomics, and the diagnosis of disease. N Engl J Med 381: 64–74. [crossref]
- Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C et al. (2007) Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 315: 848–853. [crossref]
- International HapMap Consortium (2003) The international HapMap project. Nature 426: 789–796. [crossref]
- Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, et al. (2007) International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature 449: 851–861. [crossref]
- Hirschhorn JN, Daly MJ (2005) Genome-wide association studies for common diseases and complex traits. Nat Rev Genet 6: 95–108. [crossref]
- Roberts R (2008) Personalized Medicine: A Reality within this Decade. J Cardiovasc Trans Res 1: 11–16. [crossref]
- McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R et al. (2007) A common allele on chromosome 9 associated with coronary heart disease. Science 316: 1488–91. [crossref]
- Sladek R, Rocheleau G, Rung J, Dina C, Shen L et al. (2007) A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445: 881–885. [crossref]
- Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T et al. (2007) A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science 316: 1491–1493. [crossref]
- Wellcome Trust Case Control Consortium (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447: 661–678. [crossref]
- Shen GQ, Li L, Rao S, Abdullah KG, Ban JM et al. (2008) Four SNPs on chromosome 9p21 in a South Korean population implicate a genetic locus that confers high cross-race risk for development of coronary artery disease. Arterioscler Thromb Vasc Biol 28: 360–365. [crossref]
- Preuss M, König IR, Thompson JR, Erdmann J, Absher D et al. (2010) Design of the Coronary Artery Disease Genome-Wide Replication and Meta-Analysis (CARDIoGRAM) Study: A Genome-wide association meta-analysis involving more than 22 000 cases and 60 000 controls. Circ Cardiovasc Genet 3: 475–483. [crossref]
- Roberts R (2015) A genetic basis for coronary artery disease. Trends Cardiovasc Med 25: 171–178. [crossref]
- Erdmann J, Kessler T, Munoz Venegas L, Schunkert H (2018) A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc Res 114: 1241–57. [crossref]
- Assimes TL, Roberts R (2016) Genetics: implications for prevention and management of coronary artery disease. J Am Coll Cardiol 68: 2797–2818. [crossref]
- Musunuru K, Kathiresan S (2019) Genetics of common, complex coronary artery disease. Cell 177: 132–145. [crossref]
- Schunkert H, von Scheidt M, Kessler T, Stiller B, Zeng L (2018) Genetics of coronary artery disease in the light of genome-wide association studies. Clin Res Cardiol 107: 2–9. [crossref]
- Goldstein BA, Knowles JW, Salfati E, Ioannidis JP, Assimes TL (2014) Simple, standardized incorporation of genetic risk into non-genetic risk prediction tools for complex traits: coronary heart disease as an example. Front Genet 5: 254 [crossref]
- Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR et al. (1995) Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland coronary prevention study group. N Engl J Med 333: 1301–1307. [crossref]
- Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A et al. (2004) Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the interheart study): case-control study. Lancet 364: 937–952. [crossref]
- Downs JR, Clearfield M, Weis S, Whitney E, Shapiro DR, et al. (1998) Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 279: 1615–1622. [crossref]
- Kones R, Rumana U (2015) Current treatment of dyslipidemia: a new paradigm for statin drug use and the need for additional therapies. Drugs 75: 1187–1199. [crossref]
- Fujita H, Okada T, Inami I, Makimoto M, Hosono S et al. (2008) Low– density lipoprotein profile changes during the neonatal period. J Perinatol 28: 335–340. [crossref]
- Pac-Kozuchowska E (2007) Evaluation of lipids, lipoproteins and apolipoproteins concentrations in cord blood serum of newborns from rural and urban environments. Ann Agric Environ Med 14: 25–29. [crossref]
- Ference BA, Yoo W, Alesh I, Mahajan N, Mirowska KK et al. (2012) Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a Mendelian randomization analysis. J Am Coll Cardiol 60: 2631–2639. [crossref]
- Cholesterol Treatment Trialists’ (CTT) Collaboration, Baigent C, Blackwell L, Emberson J, Holland LE (2010) Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 376: 1670–1681. [crossref]
- Navar-Boggan AM, Peterson ED, D’Agostino RBS, Neely B, Sniderman AD et al. (2015) Hyperlipidemia in early adulthood increases long-term risk of coronary heart disease. Circulation 131: 451–458. [crossref]
- Mega JL, Stitziel NO, Smith JG, Chasman DI, Caulfield M et al (2015) Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet 385: 2264–71.
- Natarajan P, Young R, Stitziel NO, Padmanabhan S, Baber U et al. (2017) Polygenic risk score identifies subgroup with higher burden of atherosclerosis and greater relative benefit from statin therapy in the primary prevention setting. Circulation 135: 2091–2101 [crossref]
- Inouye M, Abraham G, Nelson CP, Wood AM, Sweeting MJ, Dudbridge F, et al. (2018) Genomic risk prediction of coronary artery disease in 480,000 adults. J Am Coll Cardiol 72: 1883–189. [crossref]
- Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C et al. (2018) Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 50: 1219–1224.
- Khera AV, Emdin CA, Drake I, Natarajan P, Bick AG et al. (2016) Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med 375: 2349–2358.
- Tikkanen E, Gustafsson S, Ingelsson E 2018) Associations of fitness, physical activity, strength, and genetic risk with cardiovascular disease: longitudinal analyses in the UK Biobank study. Circulation 137: 2583–2591.