Introduction
The pathophysiology of schizophrenia is not completely understood, but most hypotheses center around the core philosophy that schizophrenia is subject to the influence of neurotransmitters, specifically dopamine [1, 2]. While traditional psychotropic agents used to treat psychiatric illness correct neurotransmitter dysfunction to alleviate symptoms, there is growing evidence to suggest that inflammation, oxidative stress, and glutamate pathological changes have an effect in psychiatric conditions.
In the glutamate hypothesis of schizophrenia, abnormal glutamate uptake by glial cells can result in decreased N-methyl-D-aspartate (NMDA) receptor function [1, 3–7]. Dysfunction of NMDA receptors results in the disruption of downstream dopamine signaling. Specifically, hypofunction of the NMDA receptors in the mesolimbic dopamine pathway can result in hyperactivation of neurons, which can present as hallucinatory symptoms. Whereas, in the mesocortical pathway, NMDA receptor hypofunction can lead to negative symptoms, including anhedonia and impaired cognition. The affinity of glutamate for the NMDA receptor can be influenced by NMDA cofactors glycine and glutathione.
Modulation of the NMDA receptor via allosteric binding of glycine can enhance glutamate binding [1, 3–4]. Sarcosine (N-methyl glycine) is a type 1 glycine transporter inhibitor (GlyT1), that increases the synaptic concentration of glycine by preventing its reuptake by glial cells. Increased glycine in the synapse is proposed to augment glutamate binding at the NMDA receptor, in theory, alleviating the many symptoms of schizophrenia.
It is also thought that the abnormal metabolism of neurotransmitters in patients with schizophrenia can consequently result in oxidative stress and damaged neurons [5]. N- acetylcysteine (NAC) is thought to relieve oxidative stress by replenishing glutathione levels to prevent neurodegenerative effects and further cognitive dysfunction [5, 8–9]. NAC increases glutathione levels by delivering cysteine to the brain, which is necessary for glutathione synthesis. Similar to the cofactor glycine, increased glutathione levels will also augment glutamate binding at the NMDA receptor.
While current approved treatment options for
neuropsychiatric disorders, including schizophrenia, have
substantial documented efficacy, there are instances in which
response to these treatment options is suboptimal [10]. In these situations, patients can be further diagnosed with treatment-resistant schizophrenia. Complementary and alternative medicine (CAM) is a treatment intervention not approved by the Food and Drug Administration (FDA), however offers additional options When other treatments Fail. Both sarcosine and NAC are recognized as CAM therapy options. This report will discuss the efficacy and safety of adjunctive sarcosine and NAC in the treatment of treatment-resistant schizophrenia.
Current Literature Evaluating Sarcosine
Recent literature has evaluated the efficacy of sarcosine in the treatment of schizophrenia. In an open-label, preliminary trial by Amiaz et al., sarcosine was initiated in 22 patients with schizophrenia. To be included, subjects had to be stabilized on an antipsychotic regimen for at least four weeks prior to the addition of sarcosine. The antipsychotic agents patients were receiving included risperidone
(n = 7), quetiapine (n = 4), zuclopenthixol intramuscular (IM) injection (n = 4), olanzapine (n = 3), fluphenazine IM injection (n = 2) and paliperidone (n = 2). Five of the 22 patients received sarcosine 2g/day, while 17 patients received 4g/day. Significant improvement from baseline was noted on the positive symptoms subscale of Positive and Negative Syndrome Scale (PANSS) following eight days of treatment (p = 0.007). Significant improvement was also seen on general psychopathology subscale of PANSS (p = 0.003). However, no significant improvement was detected in Clinical Global Impression Severity of Illness scale (CGI-S) (p = 0.08) and total PANSS score (p = 0.06) following treatment. The recruitment of this primary study was terminated following a documented safety issue reporting sarcosine may be linked to prostate cancer progression. This study was limited by the small sample size and short observation period.
A previous double-blind trial by Tsai et al. evaluated 38 patients with schizophrenia who received either adjunctive sarcosine or placebo in addition to their current antipsychotic therapy regimen for 6 weeks [12]. Patients were required to have been stabilized on their antipsychotic regimen for at least three months prior to enrollment in the study. The antipsychotic therapy regimens received included risperidone (n = 20), sulpiride (n = 6), haloperidol (n = 5), chlorpromazine (n = 1), fluphenazine decanoate (n = 1), trifluoperazine (n = 1), etumine (n = 1), sulpiride combined with chlorpromazine (n = 1), pipotiazine combined with chlorpromazine (n = 1) and medication free (n = 1). Significant improvements in positive (p < 0.0001), cognitive (p < 0.0001), and general psychiatric (p = 0.0002) symptom subscales of PANSS were observed in patients adjunctively treated with sarcosine when compared to placebo. Significant improvement was also found in Scales for the Assessment of Negative Symptoms (SANS) (p < 0.0001) and Brief Psychiatric Rating Scale (BPRS) (p = 0.0001) in patients receiving sarcosine therapy. When risperidone with adjunctive sarcosine was analyzed separately, similar results were found, as significant improvement was noted in PANSS, SANS and BPRS scales.
Lane et al. (2005) evaluated sarcosine use in patients experiencing acute exacerbations of schizophrenia [13]. It was difficult to derive clinically significant conclusions due to the numerous limitations of this study. However, improvement in general psychiatric symptoms, depression, and possibly negative symptoms was observed. Lane et al. (2008) assessed sarcosine use in 20 patients with schizophrenia as monotherapy as opposed to adjunctive therapy[14]. Compared to patients who received sarcosine 1g/day, patients who received sarcosine 2g/day experienced a 20% or greater reduction in total PANSS score. A follow-up double-blind trial, also conducted by Lane et al. (2010), randomized 60 patients with schizophrenia to receive sarcosine 2g/day or placebo[15]. Compared to placebo, sarcosine showed improvement in positive, negative and cognitive symptom subscales of PANSS. Improvement was also found among total PANSS (p=0.005), SANS (p=0.021), Quality of Life (QOL) scale (p = 0.025) and Global Assessment of Functioning (GAF) scale (p = 0.042). Similar to previous studies, the sarcosine treatment group had greater than a 20% reduction in total PANSS score. It is evident that adjunctive sarcosine therapy may provide significant benefit in patients with treatment-resistant schizophrenia who have exhausted traditional antipsychotic therapy options.
Current Literature Evaluating N-acetylcysteine (NAC)
A double-blind, placebo-controlled trial assessing the impact of adjunctive NAC in patients diagnosed with schizophrenia evaluated a primary outcome of improvement on PANSS score [5]. The treatment group received NAC 2g daily compared to placebo for 4 months. NAC was administered in addition to each patient’s current antipsychotic regimen. Four weeks following NAC discontinuation, the treatment group demonstrated a statistically significant improvement in PANSS total (p = 0.009), negative (p = 0.018) and general (p = 0.035) subscales. No significant improvement in PANSS positive subscale was found. A second randomized, double-blind, placebo-controlled trial assessed efficacy of NAC (up to 2g/day), in addition to risperidone (up to 6mg/ day), for 8 weeks in 42 patients with a baseline PANSS score greater than 20 [16]. A significant improvement in PANSS negative (p < 0.001) and total (p = 0.006) scales was found, but positive and general subscales lacked statistically significant improvements.
A systematic review and meta-analysis examined the efficacy and safety of adjunctive NAC in patients with schizophrenia among three randomized controlled trials [17]. Adjunctive NAC (2 to 6g/day) significantly improved total psychopathology (p = 0.03), but not general, positive or negative PANSS subscales. There were no significant differences observed in discontinuation rates or adverse effects (drowsiness, headache, nauseas and constipation) between placebo and treatment groups, which indicates favorable tolerability. These studies conclude that there is a modest benefit observed in augmenting maintenance antipsychotic therapy with NAC.
Case Report
A 43-year-old, single, white, female patient, with borderline intellectual functioning, has been continuously hospitalized at an inpatient New York State psychiatric institute since February 2003. She had been admitted to this same facility five times prior, with her first inpatient hospitalization dating back to June 1997, when she was diagnosed with schizophrenia, paranoid type. Her symptoms include paranoia, delusional ideation, hallucinations, disorganized thoughts, poor insight, impaired judgement, irritability, agitation and aggression. The patient has a history of physical and sexual abuse, as well as polysubstance abuse with marijuana, Lysergic Acid Diethylamide (LSD) and alcohol. The patient’s father reportedly suffered from schizophrenia, and died at age 48 from myocardial infarction.
Despite multiple treatment regimens over her lengthy hospital stay, the patient continued to have episodes of severe symptoms that interfered with daily functioning, including aggression. A trial of clozapine (Clozaril) was initiated, but was discontinued due to the development of myocarditis, leaving the patient ineligible for treatment rechallenge. After subsequent failed attempts to stabilize the patient with the use of alternative antipsychotic agents, either as monotherapy or in combination, including risperidone (Risperdal), olanzapine (Zyprexa), fluphenazine (Prolixin) and quetiapine (Seroquel), the patient’s treating psychiatrist requested a trial of CAM to augment her current psychiatric medication regimen of fluphenazine (Prolixin) decanoate 75mg intramuscular injection (IM) every other week along with oral doses of olanzapine (Zyprexa) 20mg twice daily, topiramate (Topamax) 200mg twice daily and lorazepam (Ativan) 2mg four times daily (Table 1). On 12/27/12, the patient was initiated on oral doses of NAC 600mg twice daily, followed by sarcosine 1g twice daily on 1/17/13, for treatment resistant schizophrenia. The patient was additionally treated with chloral hydrate as needed (PRN) until it was removed from the market by the Food and Drug Administration (FDA) in October 2012. The patient received hydroxyzine pamoate (Vistaril) 50mg every 4 hours PRN as a replacement for the chloral hydrate (Figures 1A-C).
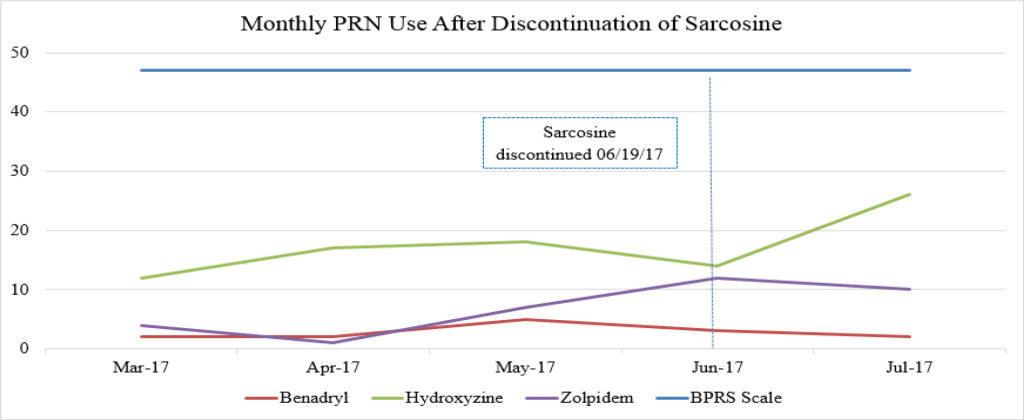
Figure 1A. Monthly PRN use before initiation of sarcosine.
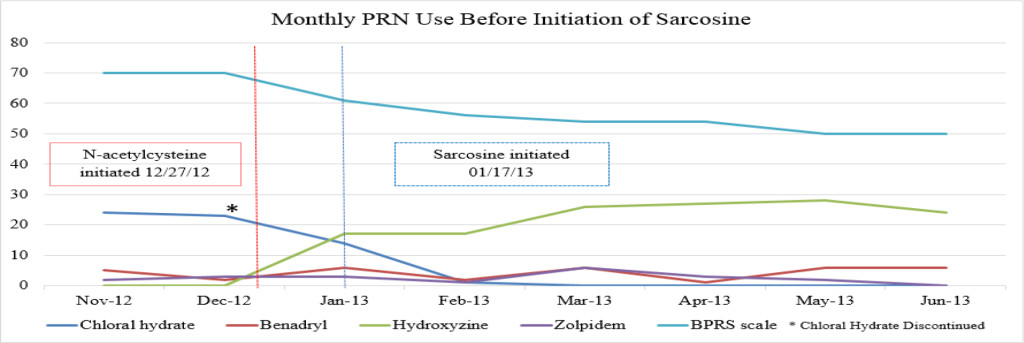
Figure 1B. Monthly PRN use after discontinuation of sarcosine.
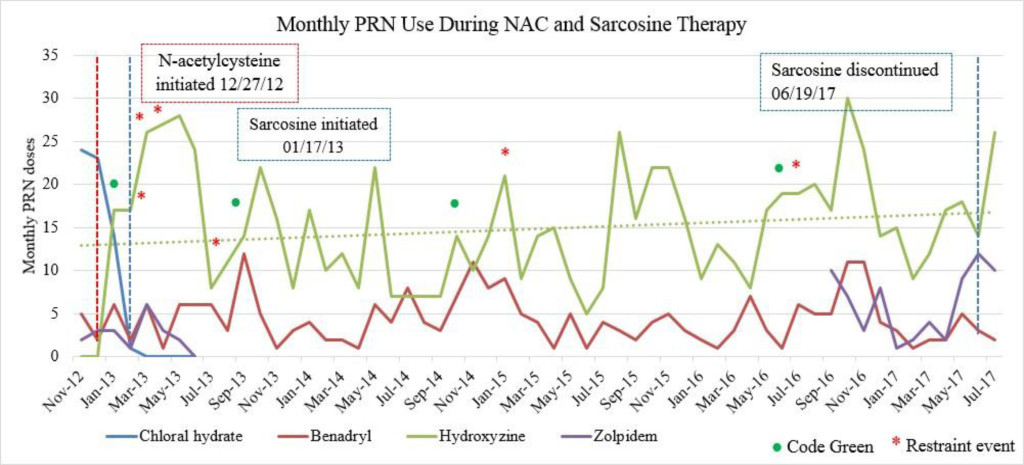
Figure 1C. Monthly PRN use during sarcosine therapy.
Table 1. Medication regimen changes over course of hospitalization.
|
Medication List 11/2/12 |
2013–2016 (date initiated) |
Medication List 4/20/17 |
|
Calcium/Vitamin D 500mg/200units BID |
Calcium/Vitamin D 500mg/200units BID |
Calcium/Vitamin D 500mg/200units BID |
|
Fluphenazine 5mg 4x daily |
Discontinued: Fluphenazine 5mg 4x daily (11/16/12) |
|
|
Fluphenazine 75mg IM injection every other week |
Fluphenazine 75mg IM injection every other week |
Fluphenazine 75mg IM injection every other week |
|
Lactulose 20mg/30mL QHS |
Lactulose 20mg/30mL QHS |
Lactulose 20mg/30mL QHS |
|
Lorazepam 2mg 4x daily |
Lorazepam 2mg 4x daily |
Lorazepam 2mg 4x daily |
|
Olanzapine 20mg BID |
Olanzapine 20mg BID |
Olanzapine 20mg BID |
|
Topiramate 200mg BID |
Topiramate 200mg BID |
Topiramate 200mg BID |
|
|
Initiated: Propranolol 20mg BID (11/2012) |
Propranolol 20mg BID |
|
|
Initiated: N-Acetylcysteine 600mg BID (12/2012) |
N-Acetylcysteine 600mg BID |
|
|
Initiated: Sarcosine 1g BID (1/2013) |
Sarcosine 1g BID |
|
|
Initiated: Doxepin 50mg 4x daily (2/2013) |
Doxepin 50mg 4x daily |
|
|
Initiated: Bisacodyl 10mg twice weekly (2/2013) |
Bisacodyl 10mg twice weekly |
|
|
Initiated: Omeprazole 20mg QAM (4/2017) |
Omeprazole 20mg QAM |
|
|
Initiated: Thiothixene 10mg 4x daily (1/2016) |
Thiothixene 10mg 4x daily |
BID=twice daily
QHS=at bedtime
QAM=in the morning
4x daily=four times daily
All doses were administered by mouth unless otherwise noted
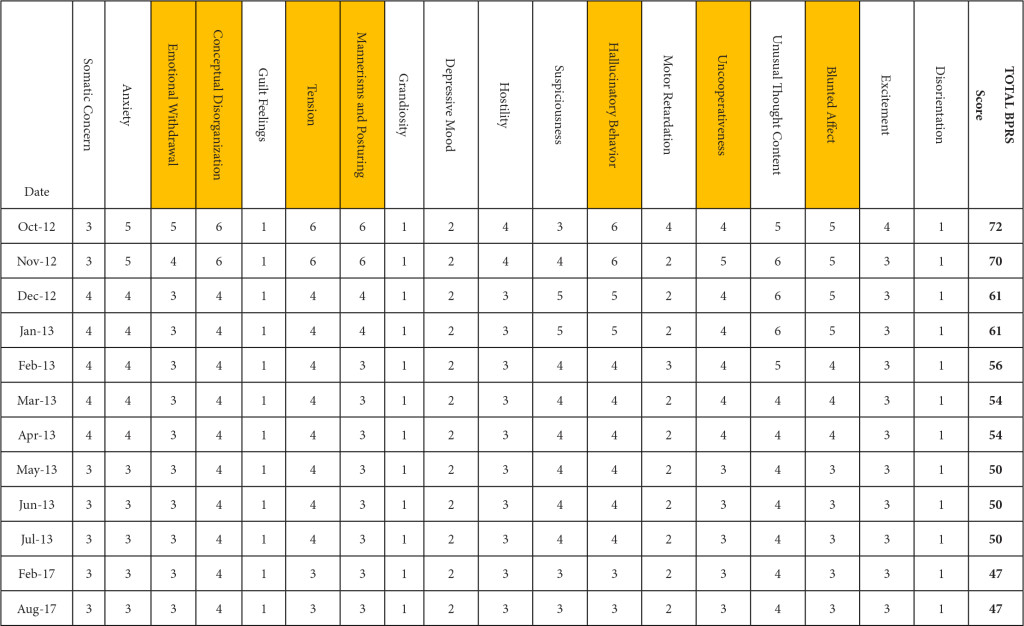
BPRS was utilized to document the progression of her treatment and the impact medication interventions had on her delusional thoughts and behaviors, including paranoia, anxiety and hostility. Prior to the initiation of NAC and sarcosine, the patient’s BPRS score was 72 in October 2012. Following the initiation of NAC and sarcosine, the patient’s BPRS score decreased, over the following 6 months of therapy, to a score of 50 in June 2013. Improvements were specifically seen in the areas of emotional withdrawal, conceptual disorganization, tension, mannerisms, hallucinatory behavior, uncooperativeness and blunted affect (Table 2).
Table 2. Improvement in BPRS score by date and domain. (highlighted boxes represent the score domains with the greatest improvements noted)
Emergent psychiatric interventions, including Code Greens (CG) and Restraint and Seclusions (R/S), along with PRN medication use, were also included in the analysis to evaluate the efficacy of the patient’s psychiatric medication regimen, as these are indicators of symptoms pertaining to agitation and aggression (Table 3). In the one year prior to the initiation of NAC and sarcosine, the patient required 2 CG (3/12/12, 4/9/12) and had 4 assaults reported (1/2/12, 3/19/12, 5/6/12, 6/11/12). She did not have a R/S. In the year following the initiation of NAC and sarcosine, the patient required 2 CG (1/4/13, 9/23/13) and had 4 assaults reported (2/25/13, 3/24/13, 4/2/13, 7/5/13), with no R/S (Figure 1C). Hydroxyzine pamoate (Vistaril) use fluctuated from month to month. Of note, doxepin (Sinequan) 50mg four times daily was initiated on 2/1/13. Her aggressive behavior, documented by reported assaults and CG, significantly decreased following her assault on 4/2/13, which is when her BPRS score was decreasing towards 50 (Tables 2 and 3). From her assault on 7/5/13 and CG on 9/23/13 until June 2017 (4 years following the initiation of NAC and sarcosine), the patient only required 2 CG (10/12/14, 6/14/16). Of note, thiothixene (Navane) 10mg four times daily was added to the patient’s medication regimen on 1/19/16.
Table 3. Emergent Psychiatric Interventions.
|
Date (1 year prior to NAC and sarcosine initiation) |
|
|
1/2/12 |
Assault |
|
3/12/12 |
Code Green |
|
3/19/12 |
Assault |
|
4/9/12 |
Code Green |
|
5/6/12 |
Assault |
|
6/11/12 |
Assault |
|
Date (Following initiation of NAC and sarcosine) |
|
|
1/4/13 |
Code Green |
|
2/25/13 (doxepin initiated on 2/1/13) |
Assault |
|
3/24/13 |
Assault |
|
4/2/13 (BPRS score decreased to 50) |
Assault |
|
7/5/13 |
Assault |
|
9/23/13 |
Code Green |
|
10/12/14 |
Code Green |
|
6/14/16 (thiothixene initiated on 1/19/16) |
Code Green |
The patient was maintained on fluphenazine (Prolixin Decanoate) olanzapine (Zyprexa), thiothixene (Navane), topiramate (Topamax), lorazepam (Ativan), hydroxyzine pamoate (Vistaril), NAC and sarcosine, until sarcosine became unavailable in April 2017. The patient was subsequently given 500mg once daily starting on 4/20/17 to conserve the remaining sarcosine supply while the facility searched for alternative sources, but was given her last dose on 6/19/17. The BPRS score closest to the time of sarcosine discontinuation was 47 on 2/29/17. On 8/1/17, nearly 2 months after sarcosine discontinuation, the BPRS score was the same (47).
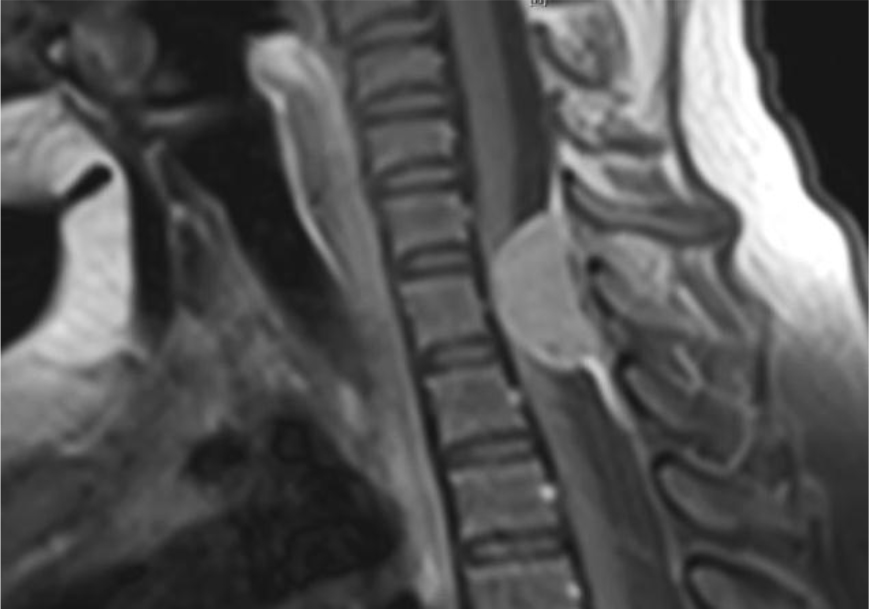
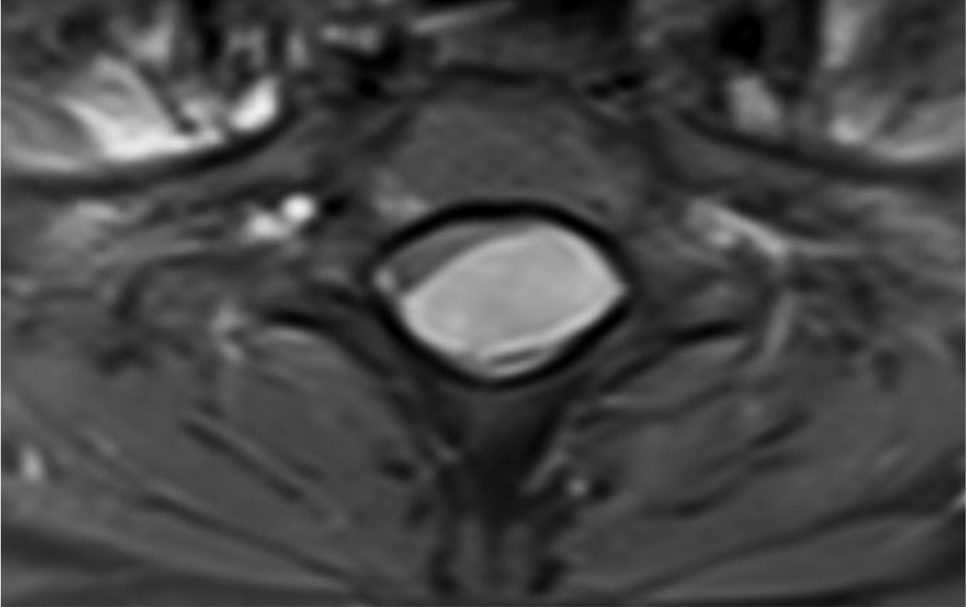
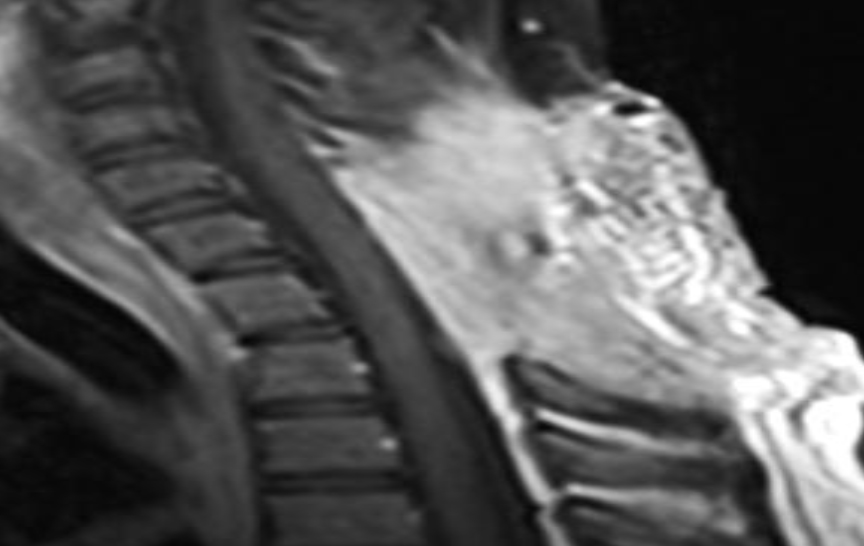
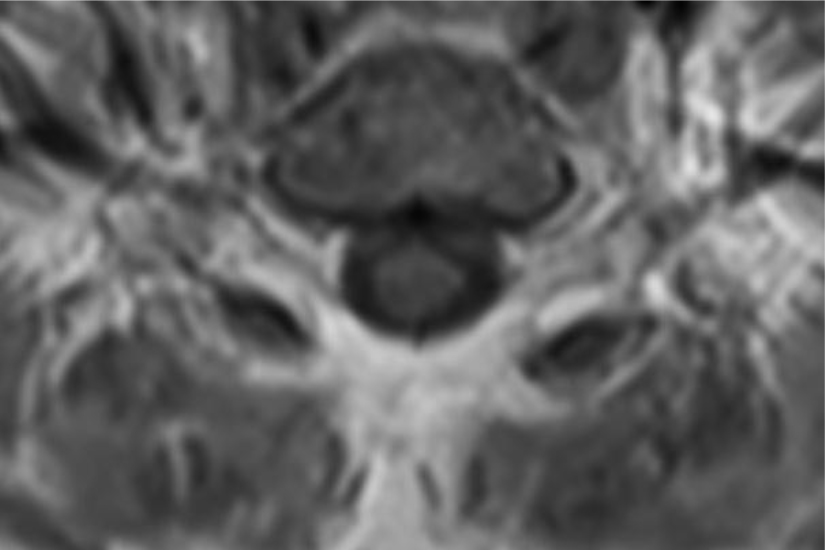
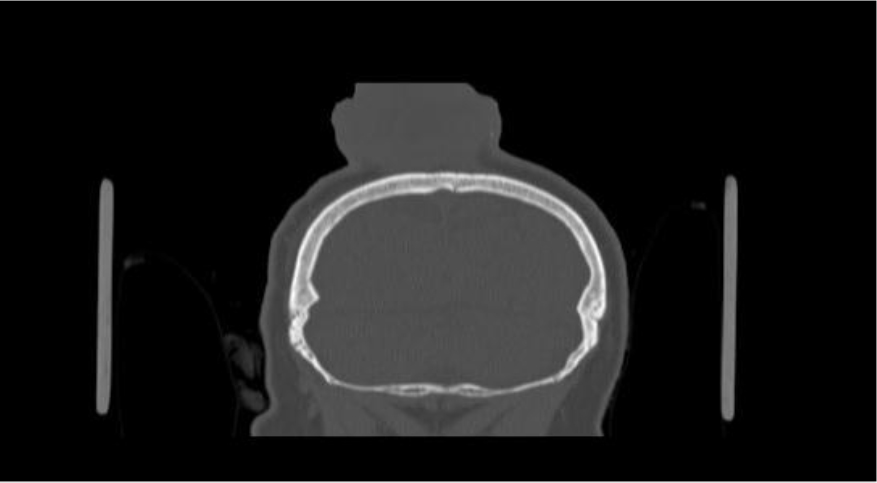
Of note, in January 2017, the patient required evaluation at an adjoining neurology clinic following ophthalmoplegia, loss of visual acuity in her right eye, and frequent headaches. The patient did present with recurrent headaches upon admission to the state psychiatric hospital. However, the frequency and severity of these headaches increased prior to her neurology consultation. An magnetic resonance imaging (MRI) scan revealed a mass extending through the orbital fissure along the cavernous sinus. The patient was subsequently diagnosed with Tolosa-Hunt Syndrome: a rare disorder characterized by non-specific inflammation in the superior orbital fissure and cavernous sinus [18]. The clinical presentation includes ophthalmoplegia and severe unilateral headaches with orbital pain. The patient received treatment with prednisone 60mg daily tapered to 15mg daily, in addition to acetaminophen (Tylenol) 650mg four times daily as needed, to alleviate her symptoms. Repeat MRI revealed minimal improvement in the mass. The patient continues to be followed by the neurology clinic with an uncertain prognosis.
Discussion
It is unknown whether symptomatic improvement can be attributed to one specific medication or a concomitant synergistic outcome. The patient’s BPRS score was report ed as 70 in November 2012. Following the initiation of NAC, the patient’s BPRS score decreased from 70 to 61 in December 2012. Following the initiation of sarcosine, the patient’s BPRS score was unchanged with a reported score of 61 in January 2012. If sarcosine were to elicit a psychotropic response, a decreased BPRS score would be expected shown as a steeper negative slope of the BPRS data line (shown in Figure 1A). The patient’s BPRS score decreased from 61 to 56 in February 2013, which could indicate a delayed response from sarcosine or be a result of doxepin (Sinequan) initiation. Following the initiation of doxepin (Sinequan) in February 2012, the patient’s BPRS score decreased from 56 to 54 in March 2013, and then continued trending downward.
There were no major fluctuations in PRN use following the initiation of NAC and sarcosine. Figure 1A shows a steep increase in hydroxyzine pamoate (Vistaril) use from December 2012 to January 2013. This was likely a result of the manufacturing discontinuation of chloral hydrate, because the patient was transitioned to hydroxyzine pamoate by the prescriber as the selected alternative to chloral hydrate. There were peaks of hydroxyzine pamoate (Vistaril) and diphenhydramine (Benadryl) use over the course of NAC and sarcosine treatment. These peaks of use tended to occur in May, and again between October and January. It is unclear what may have caused these changes in PRN use, but it may possibly be related to seasonal changes. Increased pain and discomfort, along with long-term steroid treatment may also have resulted in mood and behavioral changes leading to increased PRN use.
Sarcosine became unavailable in April 2017 due to the reclassification of sarcosine to a Category 1 substance by the FDA. Category 1 FDA designation excludes use of the agent for compounding until further efficacy and safety data is available for review [19]. The patient received her last dose of sarcosine on 6/19/17. Following the discontinuation of sarcosine, the patient’s hydroxyzine pamoate (Vistaril) use increased from 14 PRN doses in June 2017 to 26 PRN doses in July 2017. The increased PRN use following sarcosine discontinuation occurred despite the sustained lower BPRS score and reduced reports of aggressive behavior (Table 3). This could be a result of a “placebo” effect with sarcosine use or may indicate that there could have been an improvement “plateau” with sarcosine use. It could also indicate that NAC is the agent responsible for this initial and continued improvement in BPRS score. Increased risk for loss of behavioral control could have possibly been mitigated by the administration of psychiatric medications as well as the use of behavioral interventions initiated by staff. Thus decreasing the need for a CG or R/S following sarcosine discontinuation.
It is of interest that this patient was diagnosed with Tolosa-Hunt Syndrome. There are previous reports linking sarcosine to the progression of prostate and breast cancer [11, 20, 21]. Two patients diagnosed with breast cancer later presented with diffuse orbital involvement of the extraocular muscles, simulating Tolosa-Hunt syndrome [21]. Breast cancer is known to metastasize and can involve ocular structures. The patient in this case does not have a known personal or immediate family history of cancer to date. The association between sarcosine, cancer and Tolosa-Hunt syndrome is not clear, but should receive attention as the use of CAM becomes even more common.
Conclusion
There is evidence to suggest that CAM therapies, including sarcosine and NAC, can provide some therapeutic benefit to patients suffering from treatment-resistant schizophrenia who have exhausted other therapy options. The patient discussed in this case report experienced a decrease in her BPRS score following the initiation of NAC, however the magnitude of the decrease was more robust with NAC than that seen with the initiation of sarcosine. When sarcosine was discontinued there was not an increase of BPRS scores as would be expected if sarcosine were the agent responsible for her improvement.
NAC may have been the agent responsible for the patient’s symptom improvement versus a concomitant synergistic outcome with NAC and sarcosine. However, the patient is still hospitalized to date, which does question the efficacy of these alternative medication therapy options. Little is known about the long-term effects of NAC or sarcosine, when used for any patient, whether diagnosed with psychiatric illness or not. While there is no clear association, more studies pertaining to the long-term safety outcomes of NAC and sarcosine exposure should be explored before recommendations are made to promote this psychiatric therapy intervention.
References
- Yang AC, Tsai SJ (2017) New Targets for Schizophrenia Treatment beyond the Dopamine Hypothesis. Int J Mol Sci 18. crossref]
- Stahl SM, Grady MM Stahl’s (2011) Stahl’s Essential Psychopharmacology: Neuroscientific Basis and Practical Applications. (4th edn), Cambridge University Press, Cambridge, UK.
- Javitt DC, Zukin SR, Heresco-Levy U, Umbricht D (2012) Has an angel shown the way? Etiological and therapeutic implications of the PCP/NMDA model of schizophrenia. Schizophr Bull 38: 958–966. [crossref]
- Lee MY, Lin YR, Tu YS, Tseng YJ, Chan M, et al. (2017) Effects of sarcosine and N, N-dimethylglycine on NMDA receptor-mediated excitatory field potentials. J Biomed Sci 24: 18. [crossref]
- Berk M, Copolov D, Dean O, Lu K, Jeavons S, et al. (2008) N-acetylcysteine as a glutathione precursor for schizophrenia, double-blind, randomized, placebo-controlled trial. Biol Psychiatry 64: 361–368. [crossref]
- Dean O, Giorlando F, Berk M (2011) N-acetylcysteine in psychiatry: current therapeutic evidence and potential mechanisms of action. J Psychiatry Neurosci 36: 78–86. [crossref]
- Gysin R, Kraftsik R, Sandell J, Bovet P, Chappuis C, et al. (2007) Impaired glutathione synthesis in schizophrenia: convergent genetic and functional evidence. Proc Natl Acad Sci USA 104: 16621–16626. [crossref]
- Dringen R, Gutterer JM, Hirrlinger J (2000) Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur J Biochem 267: 4912–4916. [crossref]
- Chen G, Shi J, Hu Z, Hang C (2008) Inhibitory effect on cerebral inflammatory response following traumatic brain injury in rats: a potential neuroprotective mechanism of N-acetylcysteine. Mediators Inflamm 2008: 716458. [crossref]
- Lehman AF, Lieberman JA, Dixon LB, McGlashan TH, Miller AL, et al. (2004) Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry 161: 1–56. [crossref]
- Amiaz R, Kent I, Rubinstein K, Sela BA, Javitt D, et al. (2015) Safety, tolerability and pharmacokinetics of open label sarcosine added on to anti-psychotic treatment in schizophrenia – preliminary study. Isr J Psychiatry Relat Sci 52: 12–15. [crossref]
- Tsai G, Lane HY, Yang P, Chong MY, Lange N (2004) Glycine transporter I inhibitor, N-methylglycine (sarcosine), added to antipsychotics for the treatment of schizophrenia. Biol Psychiatry 55: 452–456. [crossref]
- Lane HY, Chang YC, Liu YC, Chiu CC, Tsai GE (2005) Sarcosine or D-Serine add-on treatment for acute exacerbation of schizophrenia: a randomized, double-blind, placebo-controlled study. Arch Gen Psychiatry 62: 1196–1204. [crossref]
- Lane HY, Liu YC, Huang CL, Chang YC, Liau CH, et al. (2008) Sarcosine (N-methylglycine) treatment for acute schizophrenia: a randomized, double-blind study. Biol Psychiatry 63: 9–12. [crossref]
- Lane HY, Lin CH, Huang YJ, Liao CH, Chang YC, et al. (2010) A randomized, double-blind, placebo-controlled comparison study of sarcosine (N-methylglycine) and D-serine add-on treatment for schizophrenia. Int J Neuropsychopharmacol 13: 451–460. [crossref]
- Farokhnia M, Azarkolah A, Adinehfar F, Khodaie-Ardakani MR, Hosseini SM, et al. (2013) N-acetylcysteine as an adjunct to risperidone for treatment of negative symptoms in patients with chronic schizophrenia: a randomized, double-blind, placebo-controlled study. Clin Neuropharmacol 36: 185–192. [crossref]
- Zheng W, Zhang QE, Cai DB, Yang XH, Qiu Y, et al. (2018) N-acetylcysteine for major mental disorders: a systematic review and meta-analysis of randomized controlled trials. Acta Psychiatr Scand 137: 391–400. [crossref]
- Cohn DF, Carasso R, Streifler M (1979) Painful ophthalmoplegia: the Tolosa-Hunt syndrome. Eur Neurol 18: 373–381. [crossref]
- U.S. Food and Drug Administration (2017) Update: FDA revises final guidance on interim policy for certain bulk drug substances used in compounding. Washington, D.C, USA.
- Sreekumar A, Poisson LM, Rajendiran TM, Khan AP, Cao Q, et al. (2009) Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 457: 910–914. [crossref]
- Harnett AN, Kemp EG, Fraser G (1999) Metastatic breast cancer presenting as Tolosa-Hunt syndrome. Clin Oncol (R Coll Radiol) 11: 407–409. [crossref]