Abstract
The brain is a central key organ of the body containing the second highest lipid content only after adipose tissue. Lipids as the main structural components of biological membranes play important roles in a vast number of biological processes within the brain such as energy homeostasis, material transport, signal transduction, neurogenesis and synaptogenesis, providing a balanced cellular environment required for proper functioning of brain cells. Lipids and their metabolism are of great physiological importance in view of the crucial roles of lipids in brain development and function. Astrocytes are the most abundant glial cells in the brain and involved in various processes including metabolic homeostasis, blood brain barrier maintenance, neuronal support and crosstalk. Disturbances in lipid metabolism and astrocytic functions may lead to pathological alterations associated with numerous neurological diseases like Alzheimer’s Disease (AD) recognized as the most frequent cause of dementia leading to major progressive memory and cognitive deficits as well as Glioblastoma (GBM) known as the most aggressive malignant brain tumor with a poor prognosis. Herein, we not only review the level and role of altered lipid metabolism in correlation with astrocytic function and astrocyte-neuron crosstalk in AD and GBM, but also discuss important lipid-related metabolites and proteins participating in possible mechanisms of pathologically dysregulated lipid metabolism, offering potential therapeutic targets in targeted molecular therapies for AD and GBM.
Keywords
Astrocyte, Lipid metabolism, Alzheimer’s disease, Glioblastoma
Introduction
The brain is a central and pivotal organ highly enriched in lipids (constituting 50% to 60% of brain dry weight) [1], the major biomacromolecules characterized with poor water-solubility and good solubility in non-polar organic solvent, and is regarded with the second highest lipid content next to the adipose tissue [2]. Lipids are a class of fatty substances differing in overall structure, molecular weight, head group configuration, carbon-carbon bond formation and other factors, among which fatty acids, phospholipids, sphingolipids, sterol lipids and triglycerides are the five main brain lipid classes [3], serving as basic structural components of biological membranes and participating in a broad variety of physiological events, including chemical energy generation and storage, substance transport, cellular signaling, neural differentiation, axonal regeneration, synaptogenesis, synaptic plasticity and brain development [4-14].
The brain consists of neurons and non-neuronal cells such as glial and vascular epithelial cells, of which astrocytes represent the most abundant glial cells [15,16]. Astrocytes mediate diverse biological activities under physiological conditions, including structural and energy support for neurons [17,18], neuronal development and maintenance [19,20], formation, function and plasticity of synapses [21,22], modulation of synaptic transmission [22], metabolomic homeostasis [23] as well as integrity of the Blood-brain Barrier (BBB) [24,25] which is a semipermeable membrane regulating solute exchange between blood and brain parenchyma to maintain CNS homeostasis and function and partially separating local lipid metabolism of the brain from that of the body [25-33]. Apart from the well-known enzymatic capacity of glycogenesis and glycolysis [34-38], equipment of lipid metabolism also exists in astrocytes, providing membrane components for neurons and other glial cells [39,40] and playing fundamental roles in astrocyte function including membrane fluidity, energy generation and intercellular signaling. Emerging evidence has shown that astrocytic usage of lipids stored in droplets via mitochondrial β-oxidation fulfills crucial energy-providing and neuroprotective roles in the brain [18,41], whereby disruption in lipid metabolism, structure and function of astrocytes may lead to pathogenic mechanisms underlying an array of neurological diseases.
Lipid Classification in the Brain
Fatty Acids
As one of the most well-known lipid class, Fatty Acids (FAs), the essential monomeric constituents of all lipids, account for almost 20% of the energy source through oxidation, for which astrocytes as the major provider of fatty acid β-oxidation may be the essential place [42-44]. Additionally, fatty acids can also be utilized by astrocytes for producing ketone bodies under particular conditions (e.g. ischemia), serving as a substrate for neuronal energy production-related Tricarboxylic Acid (TCA) cycle [45]. Fatty acids permeate the Blood-brain Barrier (BBB) via passive (dissociation from albumin carriers, binditheng to luminal membrane which belong to endothelia cells, ATP-independent release and entrance into the cytosol) and/or protein-mediated transport (e.g. Fatty Acid Transport Proteins (FATPs), fatty acid translocase/CD36 (FAT/CD36), Fatty Acid Binding Proteins (FABPs) and caveolin-1) [46,47]. Fatty acids can be further divided into unsaturated and saturated fatty acids, from which the former subclass contains Monounsaturated Fatty Acids (MUFAs) and Polyunsaturated Fatty Acids (PUFAs), while the latter comprises palmitic acid, stearic acid and others [48,49]. PUFAs are highly enriched in the brain, with 3- to 4-fold level over other tissues [50,51]. What’s more, essential PUFAs play key roles in brain activity and development [52,53], in which the ω-3 Docosahexaenoic Acid (DHA) are particularly involved in synaptogenesis, neurogenesis and neuroprotection in the brain [54-57].
Phospholipids
As the most abundant constituent of major categories of membrane lipids [58,59], Phospholipids (PLs) generally consist of two hydrophobic tails of fatty acids differing in length and a backbone-attached hydrophilic phosphate group [60-62].
Phospholipids, which are synthesized in the mitochondria and Endoplasmic Reticulum (ER) tracing from diacylglycerol and phosphatidic acid, spontaneously aggregate into the formation of bimolecular layers in aqueous environments on account of configuration and amphipathic property [63]. Phospholipids can be classified into glycerophospholipids and phosphosphingolipids, of which glycerophospholipids are the prominent glycerol-based class of lipid molecules which can be further subclassified into subtypes such as Phosphatidic Acid (PA), Phosphatidylcholine (PC), Phosphatidylethanolamine (PE), Phosphatidylglycerol (PG), Phosphatidylinositol (PI) and Phosphatidylserine (PS) on the basis of variation in hydrophilic head groups and participate in a variety of physiological activities in the brain [59,64,65]. Moreover, fates of brain cells are influenced by exposure to different phospholipids, such as differentiation of neural cells into astrocytes was promoted and inhibited with PE and PC treatment, respectively [66].
Sphingolipids
Sphingolipids containing sphingoid bases (also known as long-chain bases) and a set of aliphatic amino alcohols that includes sphingosine are mainly synthesized in Endoplasmic Reticulum (ER). Sphingolipids comprise a large group of lipid molecules through compounding with different functional groups, such as ceramide (functional group of single hydrogen atoms) and Sphingomyelin (SM) (functional groups including phosphocholine) with regards to structural composition, functioning as building blocks of membranes (e.g. lipid rafts) [67] and playing fundamental roles in formation and regulation of synapse structure and function [68], cell recognition, signal transmission and inflammatory regulation of astrocytes [69-72]. Besides, sphingolipid metabolites have also been discovered to exert regulatory roles in autophagy, cancer cell growth, response to DNA damage and inflammation [73-75].
Sterol Lipids
Sterol lipids include numerous organic molecules, of which cholesterol with four hydrocarbon rings is the main part. Cholesterol can be synthesized in ER by all nucleated cells, while over 70% of total body cholesterol are provided by the diet [76], namely the cholesterol absorbed in the gut transfers into the liver and then spreads through the body. What is noteworthy is that the brain, unlike other organs, makes its own cholesterol because of effective prevention of peripheral cholesterol exchange between brain tissue and plasma cholesterol-carrying lipoproteins by the BBB [77-79]. In brain tissue, de novo synthesis of cholesterol is mainly performed in astrocytes which are considered as the main cholesterol producer in the brain [80], though the majority of sterol is synthesized in oligodendrocytes in developing brain and has an association with myelination [81] and oligodendrocytes, besides, cholesterol can also be synthesized in many other cell types [82-84]. Apart from de novo synthesis [85], brain cells are able to acquire cholesterol from neighboring cells through the absorption of cholesterol-laden lipoproteins (e.g. Apolipoprotein E (APOE)) in a receptor-mediated way [86,87], in which lipoprotein synthesis for cholesterol transport occurs in astrocytes [88]. With abundant existence in myelin and lipid membranes [81], cholesterol fulfills vital roles in the brain, including BBB integrity, organization of lipid rafts (discrete microdomains present in the external leaflet of plasma membrane), regulation of cell membrane flexibility (through interaction with neighbouring phospholipids) and localization and activity of diverse membrane proteins (e.g. membrane receptor and transporter proteins), axonal guidance, formation and maintenance of synapses and dendrites, synaptic membranerelated fluidity and ion channel function, glucose transport, intracellular signaling and other important neuronal functions [84,89-103].
Triglycerides
As the major form of FA deposition and the optimal form of FA triesters of glycerol, Triglycerides (TGs) are essential ingredients of glycerolipid synthesis by assembling with other glycerol molecules [104]. Triglycerides mainly generated in the adipose tissue and liver can reach other tissues with the package into lipoproteins containing a hydrophilic exterior and a hydrophobic lipid core, including chylomicrons, Very-lowdensity Lipoproteins (VLDL), low-density lipoproteins (LDL), very-high-density lipoproteins (VHDL) and high-density lipoproteins (HDL) only which can cross the BBB [105-108]. Additionally, apolipoprotein E (ApoE) and apolipoprotein J (ApoJ), the most abundant apolipoproteins synthesized in astrocytes, serve as receptor ligands on HDL [109-111] and play fundamental roles in lipid metabolism-associated structural support, enzyme activity and substrate delivery [110,112-114].
Astrocyte-Neuron Coupling of Lipid Metabolism
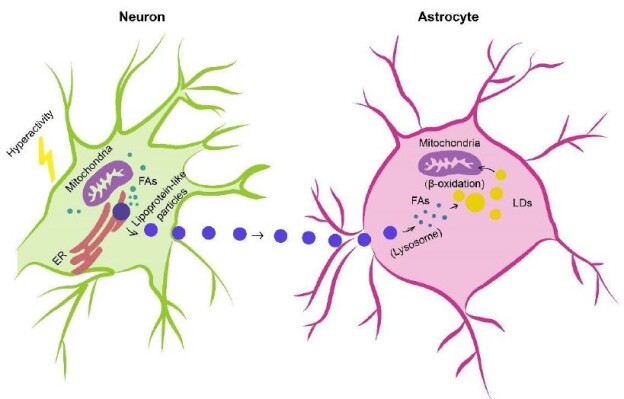
In humans, the brain representing, on average, merely 2% of total body weight consumes approximately and over 20% of energy substrates during quiet waking and diverse tasks, respectively [115,116], which depends on relatively efficient metabolic coupling between astrocytes and neurons. Physiologically, astrocytes are considered primarily as glycolytic cells with a large enzymatic capacity for glycolysis [115,117,118], whereas neurons are predominantly oxidative [119-121]. Besides the glucose metabolism in which astrocytes participate in the delivery of blood-derived glucose to neurons as an obligatory energy fuel, glycogen storage and activitydependent L-lactate production as a metabolic substrate for neurons during aerobic glycolysis [115,122-124], astrocytes-neuron coupling of lipid metabolism has also been suggested to occur as a response to neuronal activity in protection of neurons from lipotoxicity [125,126]. This is a mechanism proposing that L-lactate-derived de novo synthesis of free fatty acids (FFAs) in overstimulated neurons is triggered during astrocyte-neuron L-lactate shuttle (ANLS), resulting in excess FFAs in association to lipotoxicity-related reactive oxygen species (ROS) and lipid peroxidation chain reaction [127], peroxidized FAs with devastating effects [127] are then transferred from hyperactive neurons to astrocytes via apolipoprotein E-positive lipid particles, where they are directly stored in lipid droplets (LDs) [125,126,128] which are dynamic organelles possessing a core of neutral lipids (e.g. cholesterol esters (CEs) and triacylglycerides (TAGs)), influencing fatty acid breakdown for energy production [129] and buffering excess FFAs to prevent lipid accumulation [130] as well as utilized as an energy substrate in β-oxidation [126] (Figure 1).
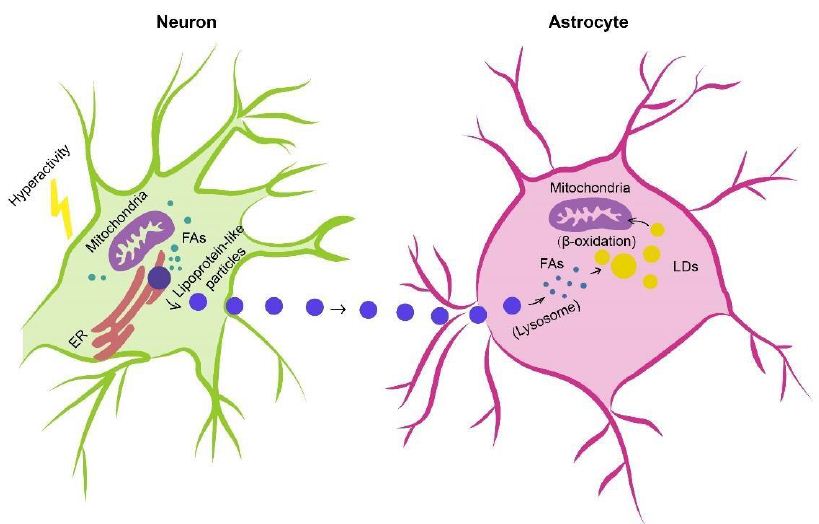
Figure 1: Astrocyte-Neuron coupling of lipid metabolism. Excess fatty acids produced in hyperactive neurons are transferred via lipid particles associated with APOE to astrocytes, where fatty acids are delivered to lipid droplets after endocytosis of neuron-derived lipid particles, detoxified as a means of neuron protection under conditions of enhanced activity as well as consumed by mitochondrial oxidation (e.g. β-oxidation). FAs, fatty acids; APOE, apolipoprotein E; LDs, lipid droplets.
AD
With the worldwide increase in longevity, Alzheimer’s disease (AD) as the most common form of senile dementia is rapidly becoming a major health problem [131,133]. AD is a devastating irreversible neurodegenerative disease clinically defined by memory loss, neuropsychiatric abnormalities, cognitive impairment, behaviour deficits and progressive decline of self-care capacity [134-136] as well as pathologically characterized by extracellular amyloid-ß (Aβ) plaques and intracellular neurofibrillary tangles (NFTs) composed of hyperphosphorylated microtubuleassociated protein tau [137-139]. Moreover, accumulation of lipid granules in glia, besides notorious Aβ deposition and tau aggregates, was noticed with the examination of Auguste Deter’s brain (the first described AD patient), initially establishing a possible involvement of perturbations of lipid metabolism in AD pathology [140,141]. Altered lipid metabolism has also been further described with important roles in AD pathogenesis [142-151].
Recent AD pathology-related lipidome studies have demonstrated changes in content of numerous lipids (Table 1). Substantial differences in fatty acid levels were observed in AD brain tissues [152,153], including a decrease in levels of docosahexaenoic acid (DHA) present in frontal cortex gray matter [154] and hippocampus [155] to which damage correlates with impaired learning and memory [156], suggesting a dysregulation of fatty acid metabolism and may potentially marking this neurodegenerative disease [157]. Cholesterol accumulation observed in senile plaques and influenced brain regions from AD patients [158] has been reported in association with region-specific synapse loss [159]. A causal relationship between hypercholesterolemia and dysfunctional cholinergic system, cognitive impairments and pathology of amyloid and tau protein has been also demonstrated [160,161], further supporting important roles of disturbed cholesterol metabolism in AD. What’s more, detection of elevated cholesterol esters was performed in lipid raft-like mitochondria-associated ER membranes (MAMs) [162] of which hyperactivity leads to cholesterol retention and synapse loss and correlates with cognitive deficits [163] and in which accumulated cleaved products of Amyloid Precursor Protein (APP) cause mitochondrial dysfunction, interruption of cellular lipid homeostasis and membrane lipid alterations generally observed in AD pathogenesis [164,165]. Mitochondrial dysfunction, accompanied with increased oxidative stress, in neurons induces a lipid transfer to nearby astrocytes in which lipid droplets accumulate, in turn, mitochondrial dysfunction in glial cells can be caused by accumulation of peroxidated lipids and oxidative stress, contributing to neurodegenerative processes [166-168]. Growing evidence has supported nonnegligible roles of phospholipids and sphingolipids in AD pathogenesis and progression, with studies reporting that phospholipids and sphingolipids, together with acylglycerols, fatty acids and sterol lipids, present significant content changes in AD brain tissues [154,169-175].
Table 1: Summary of lipid changes in AD
| Lipids | Tissue | Changes in AD | Ref | ||
| Fatty acids | Omega-3 fatty acids | DHA | Brain; CSF; Circulation | ↓ | [176-180] |
| MFG | ↑ | [179] | |||
| FCx | ↓ | [181] | |||
| EPA | Brain; Circulation | ↓ | [180] | ||
| MFG | ↓ | [179] | |||
| DPA | Brain | ↑ | [182] | ||
| ALA | Plasma | ↑ | [183] | ||
| Omega-6 fatty acids | AA | Brain; CSF | ↑ |
[177,184,1 85] |
|
| MFG | ↓ | [179] | |||
| HPC | ↓ | [186] | |||
| LA | Brain; Plasma | ↓ | [179,187] | ||
| Saturated fatty acids | Brain; CSF | ↑ | [176] | ||
| Eicosanoids | PG | Brain | ↑ | [188] | |
| Phospholipids | Phosphatidylcholine (PC) | Total PC lipids | Brain | ↓ | [189] |
| PC-EPA | CSF | ↓ | [190] | ||
| PC-DHA | Plasma | ↓ | [191] | ||
| PC-EPA | Plasma | ↓ | [191] | ||
| Phosphatidylethanolamine (PE) | Total PE lipids | HPC | ↓ | [186] | |
| PE-SA | HPC | ↓ | [192] | ||
| PE-OA | HPC | ↓ | [192] | ||
| PE-AA | HPC | ↓ | [192] | ||
| PE-DHA | HPC | ↓ | [192] | ||
| Phosphatidylserine (PS) | Total PS lipids | Occipital lobe; Inferior parietal | ↓ | [193] | |
| lobule | |||||
| Sphingolipids | Ceramides (CM) | Total CM lipids | Brain | ↑ | [194] |
| Sphingomyelin (SM) | Total SM lipids | CSF | ↓ | [195] | |
| Triglycerides | Total TG lipids | Serum | ↓ | [196] | |
| Polyunsaturated TG | Brain | ↓ | [197] | ||
| Sterol lipids | Cholesterol | Brain | ↓ | [198] | |
| Cholesterol precursors | Brain | ↑ | [198] | ||
| Total oxidized cholesterol | Brain | ↑ | [199] |
PC, phosphatidylcholine; PE, phosphatidylethanolamine; PS, phosphatidylserine; CM, ceramides; SM, sphingomyelin; TG, triglyceride; AA, arachidonic acid; ALA, alpha-linolenic acid; DHA, docosahexaenoic acid; DPA, docosapentaenoic acid; EPA, eicosapentaenoic acid; LA, linoleic acid; OA, oleic acid; SA, stearic acid; PG, prostaglandin; CSF, cerebral spinal fluid; FCx, frontal cortex; HPC, hippocampus; MFG, medial frontal gyrus;↑; increased from control ↓; decreased from control.
APOE
In comparison with early-onset familial AD (EOFAD), late-onset AD (LOAD) accounts for approximately 95% of all AD cases [200,201], in which genetic predisposition, after aging, plays major roles in the onset of AD. As the strongest risk factor for LOAD, apolipoprotein E (APOE) is the main lipoprotein in the brain and plays pivotal roles in brain lipid metabolism, membrane remodelling and neuronal growth and repair [202-206]. APOE mainly produced by astrocytes is released into extracellular space where essential lipids (e.g. cholesterol) are delivered to neurons adopting APOE-bound cargo through APOE receptors expressed on the neuronal surface [202]. In addition to the capacities of Aβ binding and influencing Aβ aggregation and clearance [204,207], APOE participates in indirect regulation of Aβ metabolism through interactions with receptors (e.g. low-density lipoprotein receptorrelated protein 1 (LRP1)) [206,208-213]. Critical and isoform-specific role of APOE has also been demonstrated in formation of intraparenchymal Aβ deposits in amyloid precursor protein (APP) transgenic mice [214-217]. APOE exists with 3 different alleles namely APOEε2, APOEε3 and APOEε4, translating to 3 protein isoforms termed APOE2, APOE3 and APOE4, of which APOE4 present in approximately 14% of worldwide populations [205,218] is the most prevalent genetic risk factor for AD [219-222]. A single amino acid difference between APOE3 and APOE4 (Cys 112 Arg) brings about a conformational change influencing the binding to Aβ, lipids and apolipoprotein receptors [223]. APOEε2 considered as a protective genetic factor associated with reduced risk for AD and late age at onset [219,224] has been reported to orchestrate differences in lipidome and transcriptome profiles of postmortem AD brain [218,225]. Conversely, APOEε4 markedly elevates AD risk [219,224], in which heterozygous and homozygous APOEε4 allele may increase AD risk by 3 and 12 times, respectively [223], accelerates disease course and worsens brain pathology [226-228]. A correlation between APOE4 genotype and increased expression of Serpina3n, a gene expressed by astrocytes and considered as a strong marker of reactive and aged astrocytes in the brain [229,230], has been reported with a possible contribution to the pathogenic role of APOE4 in AD [231]. Higher APOE4 level in Cerebral Spinal Fluid (CSF) of AD patients compared with that of control individuals has been connected to accelerated Aβ oligomer accumulation [232]. APOE4 may retard Aβ clearance and favour Aβ deposition via binding to Aβ after specific fragmentation [205,223]. APOE4 was reported to trap ATP-binding cassette transporters A1 (ABACA1) (a regulator of APOE4 lapidation in protection from lipidpoor ApoE4 aggregation) in late rather than recycling endosomes and alter ABACA1 membrane trafficking in astrocytes, which might result in reduced Aβ degradation [233]. Insufficient Aβ clearance also affects accumulation in synaptic cleft, contributing to disruption of hippocampal long-term synaptic plasticity related to learning and memory abilities [234]. APOE4 is internalized in APOE receptors such as low-density lipoprotein receptor-related protein 1 (LRP1) which is also a member of Aβ receptors including very low-density lipoprotein receptor (VLDLR) and apolipoprotein E receptor 2 (APOER2) [209]. Additionally, APOE4-induced reduction of dendritic spine density in mice [234,235] is consistent with pathological changes (dendritic spine density reduction and synapse loss) observed in brain tissues from AD APOEε4-carriers [236]. APOE4 causes widespread AD phenotypes-associated cellular and molecular alterations in brain cells derived from human induced pluripotent stem cells (iPSCs), among which increased Aβ secretion as well as impaired Aβ uptake and cholesterol accumulation occurred in neurons and astrocytes, respectively [237]. Astrocytic lipid metabolism is influenced by APOE4 [237,238], in which increased fatty acid unsaturation and lipid droplet (LD) accumulation were found in APOE4-expressing human iPSC-derived astrocytes, which can be restored to basal state through supplementation of culture medium with choline (a soluble phospholipid precursor) [238]. Furthermore, APOE4 can also impair astrocyte-neuron coupling of fatty acid metabolism via decreased fatty acid (FA) sequestering in LDs, reduced LD transport efficiency and lowered FA oxidation, resulting in lipid accumulation in astrocytes and hippocampus, diminished abilities of astrocytes in neuronal lipid elimination and FA degradation, accelerated lipid dysregulation and increased AD risk [239].
ACSBG1 and ACSL6
Cellular accumulation and activation of fatty acids (FAs) either synthesized de novo or taken up from diets require the ATP-dependent reaction catalyzed by acyl-CoA synthetases (ACSs), a family of enzymes initiating FA metabolism-related reactions through ligation to coenzyme A (CoA) [240]. ACS enzyme family contain various members differing in distribution and fatty acid substrate preference [241], among which only two show specific enrichment in the brain, ACSBG1 and ACSL6 [242,243], suggesting their potentially particular roles in modulation of brain fatty acid metabolism. ACSBG1, almost exclusively expressed in astrocytes, have preferences for a wide range of substrates containing long-chain saturated and unsaturated fatty acids [244,245]. ACSBG1 knockdown in vitro results in decreased ACS enzymatic activity and FA oxidation [245], indicating its participation in astrocytic FA oxidation, however, clear roles of ACSBG1 in brain function and/or dysfunction still remain poorly understood. ACSL6 showing high expression in the brain was reported to be downregulated in age-related neurodegenerative diseases [246,247] and in direct correlation with neurite outgrowth [248-252]. With high substrate preference for docosahexaenoic acid (DHA) of which low levels are associated with AD pathophysiology [253], ACSL6 has been revealed with key roles in regulating DHA incorporation into neuronal membranes using Acsl6 deficient mice with significant reduction in DHA-containing phospholipids and impaired memory [254,255]. Critical roles of ACSL6 in brain DHA retention and neuroprotection are further supported by findings that ACSL6 depletion led to markedly reduced levels of brain membrane phospholipid DHA, spatial memory deficits, hyperlocomotion, increased cholesterol biosynthesis and age-related neuroinflammation [256]. What’s noteworthy is that astrocyte-specific depletion had minimal influence on membrane lipid composition [256] in consideration of ACSL6 enrichment in astrocytes [240,257-261], possibly due to the expression of a DHA-nonpreferring variant [251,262-267] and enrichment of Y-gate domain rather than DHA-preferring F-gate domain in astrocytes [251].
ATAD3A
ATPase family AAA-domain containing protein 3A (ATAD3A), a nuclear-encoded mitochondrial membrane-anchored protein belonging to the AAA+-ATPase protein family and simultaneously interacting with inner and outer mitochondrial membranes, is implicated in a variety of biological processes including stability maintenance of mitochondrial DNA (mtDNA), regulation of mitochondrial dynamics and cholesterol metabolism [268-270]. ATAD3A deficiency led to neurodegenerative phenotypes in association with cholesterol elevation, downregulated expression of cholesterol metabolism-related genes [269], optic atrophy and axonal neuropathy [271]. Oligomerization and accumulation of ATAD3A at MAMs, lipid raft-like ER subdomain rich in sphingomyelin and cholesterol [272] and associated with diverse metabolic functions such as lipid metabolism, mitochondrial function and calcium homeostasis [273-277], have been discovered in both mouse models and postmortem human brain tissues of Alzheimer’s disease [278]. Aberrantly oligomerized ATAD3A leads to cholesterol accumulation via expression inhibition of cholesterol clearancemediating cytochrome P450 family 46 subfamily A member 1 (CYP46A1) located on MAMs of which deficiency correlates with cholesterol disturbance, amyloid aggregation and cognitive impairments [279], AD-like MAM hyperconnectivity (e.g. impaired MAM integrity) [277] as well as synapse loss [278]. MAM-resident cholesterol imbalance facilitates amyloidogenic APP cleavage [165], in turn, retention of APP proteolytic fragments at MAMs interrupts cholesterol trafficking and homeostasis [280]. Additionally, blocking ATAD3A oligomerization by heterozygous knockout or pharmacological inhibition treated with DA1 peptide has been reported in causal relationship with cholesterol turnover normalization, MAM integrity enhancement, APP processing suppression, synapse loss mitigation and ultimate reduction of AD-like neuropathology and cognitive impairments [278], further revealing a role of ATAD3A in AD pathology and suggesting a potential therapeutic strategy of retarding AD progression through manipulation of abnormal ATAD3A oligomerization.
FoxO3
Forkhead box O transcription factor 3 (FoxO3) belonging to the forkhead box (FOX) family sharing an evolutionarily conserved forkhead DNA-binding domain composed of 80 to 100 amino acids [281]and possessing single nucleotide polymorphisms (SNPs) associated with human longevity [282,283] functions as a mediator of biological processes promoting lifespan and preventing aging-related diseases [284,285], of which alterations are involved in carcinoma, cardiovascular and neurodegenerative diseases [283,286-289]. FoxO3 plays a pivotal role in quiescence maintenance of neural stem cells (NSCs) in the brain, removal of which induces NSC differentiation and consequent NSC pool reduction [290-293]. Apart from capacities for neuronal survival promotion or neuronal apoptosis mediation [294,295], FoxO3 has also been shown with astrocyte proliferation controlling through inhibiting inflammatory cytokines (e.g. TNF-α and IL-1β) mediating reactive astrogliosis in neurodegenerative diseases [296-299]. Conditional knockout of FoxO3 in astrocytes was reported to impair consumption of excess fatty acids [300] which are cytotoxic and destructive to mitochondrial function [301]. FoxO3 reduction in aged mice was found to be specific to the cortex rather than the hippocampus, where FoxO3 deficiency caused cortical astrogliosis and dysregulated lipid metabolism [300]. In addition, lipid dysregulation, mitochondrial dysfunction together with Aβ uptake impairment were also observed in cultured astrocytes deficient in FoxO3, which could be reversed by astrocytic FoxO3 overexpression [300], potentially supporting the concept that FoxO3 elevation in astrocytes may retard or restore cortical astrogliosis and AD-associated impairments.
GSAP
Under typical conditions, Amyloid-β (Aβ) peptides as the products of body’s cholesterol disturbance are cleaved from amyloid precursor protein (APP) which may occur in two cellular pools, namely lipid raft-associated pool preferentially favouring APP cleavage by β- and γ-secretase as well as non-raft pools where cleavage is performed by α-secretase in a non-amyloidogenic pathway [302] and rapidly eliminated to maintain normal Aβ levels [303]. γ‐secretase activating protein (GSAP) was first reported for its regulatory roles in γ-secretase activity and specificity and its significant and selective enhancement of Aβ production through interactions with γsecretase and amyloid precursor protein carboxy‐terminal fragment (APP-CTF) [304]. Significantly upregulated GSAP level has been demonstrated in both AD mouse models and postmortem brain tissues from AD patients [305-307]. Single-nucleotide polymorphisms (SNPs) at the GSAP locus have been shown association with AD diagnosis [308,309], of which one SNP was found to correlate with GSAP expression and AD risk [310]. Genetic knockdown and pharmacological inhibition of GSAP suppress Aβ generation and deposition and tau phosphorylation in AD mouse models [304,305,311]. Apart from the promotion of APP-CTF partitioning into Aβ production-favoring lipid rafts, GSAP has also been shown to be enriched in mitochondria-associated membranes (MAMs), an intracellular domain where amyloidogenic APP processing responsible for dysregulated lipid metabolism is performed [312,313]. GSAP depletion lowers APP-CTF accumulation in lipid rafts, reduces ER-mitochondrial contacts elevated in AD [313-316], and alters lipid profiles in a direction opposite to AD pathogenesis (e.g. GSAP depletion-raised levels of phosphatidylethanolamine (PE) and phosphatidylinositol (PI) showing consistent reduction in human AD brain) [310,317]. What’s more, interactions between GSAP and multiple components related to ER-associated degradation (ERAD) regulating mitochondrial function through MAM and participating in AD pathogenesis have also been revealed, further supporting crucial roles of GSAP in attenuating AD-associated pathogenic process.
Glioblastoma
Glioma as a malignant primary brain tumor originating from astrocytes or other glial cells accounts for approximately 80% of all malignant brain tumors [318], of which glioblastoma (GBM) is the most aggressive type of brain tumor known with a 5-year survival rate below 5% [319-321]. Metabolic reprogramming has been recognized as a fundamental hallmark for carcinogenesis and progression of multiple tumors including GBM [322-324], through which tumor cells meet the high-energy demands of rapid proliferation [325]. Except for the representative metabolic feature named the Warburg effect, a phenomenon in which GBM cells rely on glycolysis for energy production under oxygen-sufficient and oxygen-insufficient conditions [323,325-327], GBM cells can also be fueled by fatty acid oxidation (FAO) as an alternative crucial energy resource to meet high-energy consumption in GBM aggressiveness [328-332], of which inhibition negatively impacted GBM proliferation and progression [333]. Oxidation of fatty acids is achieved by two major pathways, namely enzymatic oxidation mediated by peroxidases (e.g cyclooxygenase (COX), cytochrome P450 (CYP450), lipoxygenase (LOX) and phospholipase A2 (PLA2)) [334] as well as nonenzymatic self-catalyzed peroxidation (Figure 2A) of which 4-hydroxynonenal (4HNE) is an end-product showing elevated expression proportional to the grade of brain tumor malignancy [335-337]. Moreover, lipid metabolism reprogramming in association with numerous pathophysiological processes such as tumor proliferation and development [338-343] has been further evidenced with the observation of large amounts of lipid droplets (LDs) in GBM [344-346] and other tumors [347-354]. Neutral lipid core of a single LD includes cholesteryl esters and triglycerides (TGs) composed of glycerol molecules with triple hydroxyl groups esterified by fatty acids [355-358]. TGs have been demonstrated to serve as an important energy reservoir for supporting GBM cell survival, in which LDs were rapidly broken down by GBM cells via autophagy, a pivotal cellular process degrading damaged organelles and protein aggregates and recycling nutrients via hydrolysis of cytoplasmic components to ultimately maintain cellular homeostasis [359-362], to release stored fatty acids for producing energy upon energetic stress like glucose deprivation (Figure 2B), in turn, inhibition of FAO or autophagy led to LD retention and significant potentiation of GBM cell death [363], suggesting that LDs may play critical roles in regulating GBM growth and limitation of LD usage might be indispensable in GBM treatment. What’s more, cholesterol metabolism in GBM is different from that in healthy brain tissues where nearly all brain cholesterol is synthesized de novo [364-366]. Contrary to normal astrocytes mainly synthesizing cholesterol from glucose or glutamine [367,369] and converting excess cholesterol to oxysterol as an endogenous ligand of liver X receptors (LXRs) to consequently trigger efflux of surplus cholesterol via ATPbinding cassette transporter A1 (ABCA1) and suppression of cholesterol uptake by low-density lipoprotein receptors (LDLRs) [370-374], GBM cells are insufficient to de novo synthesize cholesterol and thus dependent on exogenously supplied cholesterol for survival through upregulated LDLR expression [364,375] (Figure 2C), in which LXR agonists could induce GBM cell death by lowering intracellular cholesterol content via ABCA1-dependent cholesterol efflux and LDLR inhibition [364]. Additionally, intracellular cholesterol level has been revealed to be involved in resistance against GBM cell death induced by temozolomide (TMZ), a blood-brain barrier (BBB) penetrant chemotherapy agent currently used in the standard therapy for patients with GBM [376,377]. Furthermore, sphingomyelins (SMs), an important group of phospholipids in cell membranes, together with their hydrolysis by sphingomyelinases (SMase) are crucial to effects of radio- and chemotherapy [378,381]. Ceramides which are generated by SMase-mediated SM hydrolysis caused by TMZ and radiation can induce cell apoptosis [382-384], which can be evaded through conversion of ceramides to sphingosine-1-phosphate (S1P) (Figure 3) [385-387] linked to tumor grade and implicated in GBM aggressive phenotypes [383,388].
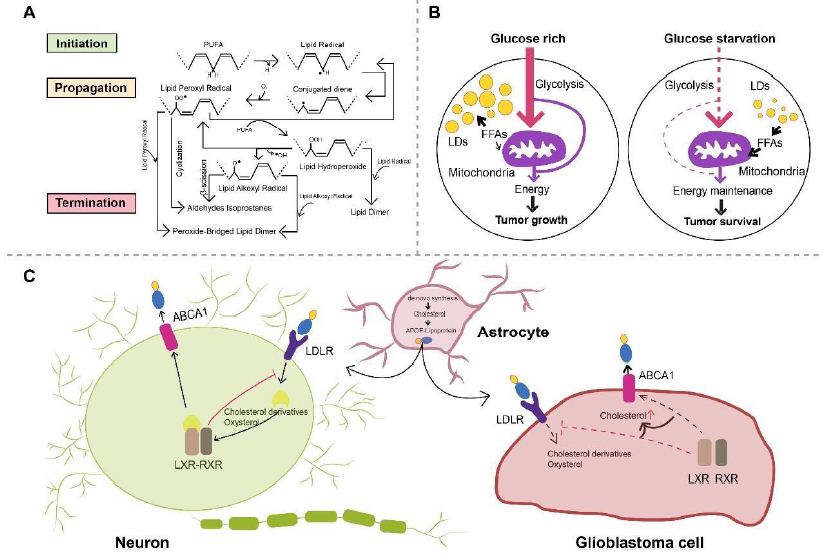
Figure 2: A. Scheme of non-enzymatic self-catalyzed lipid peroxidation. Abstraction of allylic hydrogen from PUFA induces lipid radical formation and initiates a chain reaction of lipid peroxidation, which is followed by conjugated diene-yielding molecular rearrangement. Conjugated dienes, in presence of molecular oxygen, are transformed to lipid peroxyl radical abstracting allylic hydrogen from another PUFA, forming lipid hydroperoxide and another lipid radical. Lipid hydroperoxide can be further catalyzed and transformed to lipid alkoxyl radical and lipid peroxyl radical. Lipid peroxidation is terminated when non-radical products are formed because of interaction with antioxidants. Reaction between two lipid peroxyl radicals or two lipid alkoxyl radicals will consequently form a peroxide-bridged lipid dimer, while lipid dimers can be formed by reaction between lipid hydroperoxides and lipid radicals. PUFA, polyunsaturated fatty acids. B. Schematic model of LDs hydrolysis maintaining GBM cell survival. GBM cells mainly utilize glucose to produce energy under glucose-rich conditions, while LDs can be rapidly broken down after autophagy activation upon glucose starvation, released FAs then enter mitochondria for energy production. FAs, fatty acids; LDs, lipid droplets; GBM, glioblastoma. C. Astrocytes are relied upon by neurons and GBM cells to provide de novo synthesized cholesterol. Neurons and GBM cells take up astrocyte-secreted cholesterol in APOE-containing lipoproteins. Following cholesterol uptake mediated by LDLR, oxysterol and cholesterol derivatives produced in neurons are physiological agonists for LXR of which activation leads to dimerization with RXR and subsequent elevation in ABCA1 expression. LXR activation also inhibits LDLR expression, resulting in decreased cholesterol uptake and regulating intracellular cholesterol level. On the contrary, mechanisms surveilling and regulating cholesterol are disrupted in GBM cells, in which oxysterol and cholesterol derivatives cannot activate LXR inducing intracellular cholesterol accumulation. ABCA1, ATP-binding cassette transporter A1; APOE, apolipoprotein E; GBM, glioblastoma; LDLR, low-density lipoprotein receptor; LXR, liver X receptor; RXR, retinoid X receptor.
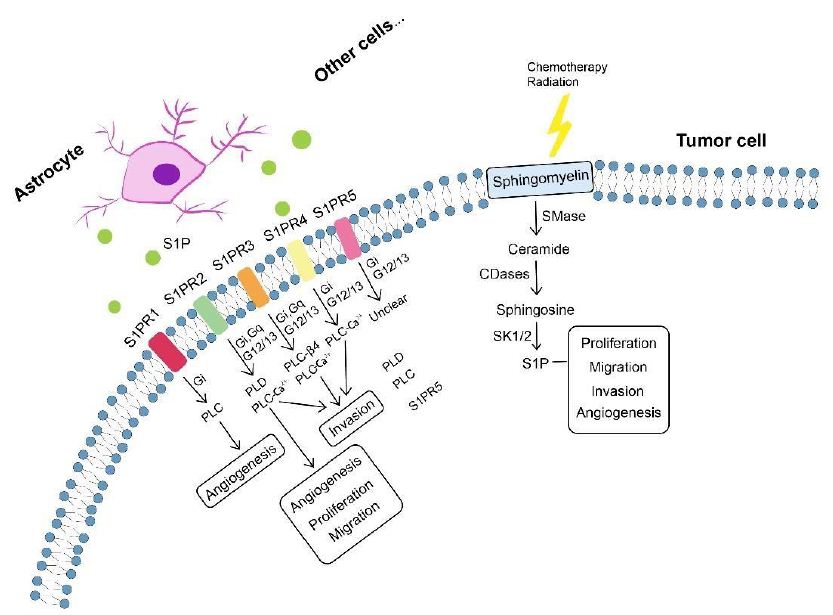
Figure 3: Sphingolipid metabolism in tumor progression. Sphingomyelin, after chemotherapy and radiation, is broken down into ceramide involved in blocking tumor progression. Ceramide can be converted by tumor cells to S1P (S1P can also be produced by astrocytes and other cells) exerting protumor effects including tumor proliferation, migration, invasion and angiogenesis. Involvement of S1P in tumor progression is specifically mediated by S1PRs (S1PR1-S1PR5) which can signal through phospholipase mechanisms. Each S1PR can couple to one or more GPCRs to signal through different phospholipases and induce phenotypes (e.g. angiogenesis, proliferation, migration and invasion). CDase, ceramidase; GPCRs, G protein-coupled receptors; SMase, sphingomyelinase; S1P, sphingosine-1-phosphate; S1PRs, S1P receptors.
S1PRs
GBM cells utilize exogenous source of S1P synthesized and exported by astrocytes and neuronal cells [389] and endogenous S1P production [390] for tumor progression. Involvement of S1P in tumor growth, migration, invasion, survival and angiogenesis [391-394] is specifically mediated by the family of G-protein coupled receptors named S1P receptors (S1PRs, S1PR1-S1PR5) [395-400]. S1PR1, S1PR2, S1PR3 and S1PR5 are expressed in human GBM cells [401-403], and elevated levels of S1PR1, S1PR2, and S1PR3 have been detected in brain tissues from GBM patients compared with healthy tissues, while only S1PR1 and S1PR2 showed significant association with GBM survival rates [401,402]. S1PRs are essential for mediating diverse S1P functions, whereas orientations in which they influence cell phenotypes still remain unclear. S1PR1 inhibition was reported to promote GBM cell proliferation, which collides with studies suggesting increased GBM proliferation by S1PR1-3, of which S1PR1 showed the strongest effects [402,404]. S1PR2 was shown to both reduce GBM migration through Rho/Rho kinase signaling pathway and participate in promoting GBM invasion [405,406]. In addition, S1PR5 has also been identified as an independent prognostic factor of GBM patients’ survival, aligning with reported role of S1PR5 in proliferation promotion [404,407]. Pharmacologically altered S1PR expression by fingolimod (FTY720), a sphingosine analogue leading to S1PR1 internalization, has been revealed to suppress astrocyte activation and change astrocytic secretion of C-X-C motif chemokine 5 (CXCL5) known to promote GBM proliferation and migration [408-410]. Furthermore, functions of individual S1P receptor subtypes are dependent upon activation of diverse downstream effector proteins, especially coupling to different G-proteins [399], such as binding of S1PR1, S1PR2 and S1PR5 with Gi, activation of Gq by S1PR2 and S1PR3 as well as signaling of S1PR2, S1PR3, and S1PR5 via G12/13 (Figure 3) [411], which alters signaling of phospholipases (particularly phospholipase C (PLC) cleaving proximal phosphodiester bonds of glycerophospholipids in production of phosphorylated headgroups and diacylglycerols [399,400]) and further activates downstream signaling molecules (e.g. extracellular signal-regulated kinase (ERK), phosphoinositide 3-kinase (PI3K) and mitogen-activated protein kinase (MEK)) (Table 2). What’s noteworthy is that a S1PR-targeted liposomal drug delivery system, named S1P/JS-K/Lipo, capable of blood-brain tumor barrier (BBTB) penetration and enhanced tumor-targeted delivery has recently been described, efficiently delivering a nitric oxide (NO) prodrug (JS-K, O2-(2,4-dinitrophenyl) 1-[(4-ethoxycarbonyl) piperazin-1-yl] diazen-1-ium-1,2-diolate) to GBM tissues via specific interactions with S1PRs highly expressed on GBM cells [412], representing a promising targeted approach for GBM therapy.
Table 2: Summary of S1PR-mediated effects in GBM
| Models | Involved S1PRs | Signaling Pathways | Findings | Ref |
| LN18 GBM cells;
U87MG GBM cells. |
S1PR1 ↑
S1PR2 ↑ S1PR3 ↑ |
PI3K/AKT1 pathway | Demonstrated association between S1P1 and S1P2 with GBM patient’s | [413] |
| survival. S1PR1/2 inhibition reduces GBM migration. | ||||
| U373MG GBM cells | S1PR1 ↑
S1PR2 ↑ S1PR3 ↑ |
MAPK/ERK and PI3Kβ pathway | S1P promotes glioma cell proliferation. | [414] |
| U373MG GBM cells; GBM6 cells; GBM12 cells. | S1PR2 | MEK1/2 and Rho/ROCK | S1P induces mRNA and protein expression of PAI-1 and uPAR, which are important for GBM invasiveness. | [415] |
| U373MG GBM cells; U118MG GBM cells. | S1PR1↑
S1PR2 S1PR3 |
MAPK-ERK
Rho/ROCK |
S1PR, S1PR2 and S1PR3 all positively contribute to S1P-stimulated glioma cell proliferation, of which S1PR1 makes the major contribution. | [416] |
| C6 glioma cells | S1PR2 | MAPK/ERK, PKC, PLC, PLD and Ca2+ signaling | S1PRs are linked to at least two signaling pathways (i.e. PTX-sensitive Gi/Go-protein pathway and toxin- insensitive Gq/G11-PLC pathway). | [417] |
| C6 glioma cells; 1321-N1 astrocytoma cells. | S1PR2 | PI3K/Cdc42/p38MAPK and PI3K/Rac1/JNK | S1PR2 mediates S1P-induced negative regulation of glioma cell migration. | [418] |
| U373MG GBM cells; U87MG GBM cells; M059K cells; U-1242 cells; A172 cells. | S1PR1 ↑
S1PR2 ↑ S1PR3 ↑ |
MAPK/ERK and PI3K | S1P potently enhances glioma cell motility by signaling through coupling of S1PRs to Gi proteins. | [419] |
| T98G glioma cells; G112 glioma cells. | S1P1, S1P2, S1P3 and S1P5 | PTEN/AKT/Egr | S1PR1 is a significant prognostic factor for glioma; | [420] |
| Downregulated S1PR1 expression increases glioma cell proliferation and enhances glioma malignancy. | ||||
| Human GBM specimens; U87 glioma cells; U251 glioma cells; T98G glioma cells; G112 glioma cells. | S1PR1↓ | Downregulated S1PR1 expression in GBM patients with a poor survival. S1PR1 signaling negatively controls glioma cell proliferation. | [421] |
AKT, v-akt murine thymoma viral oncogene homolog; Cdc42, cell division control protein 42 homolog; ERK, extracellular signal-regulated kinase; JNK, c-Jun Nterminal kinase; MAPK, mitogen-activated protein kinase; MEK, mitogen-activated protein kinase; PI3K, phosphoinositide 3-kinase; PLC, phospholipase C; PLD, phospholipase D; PTEN, phosphatase and tensin homolog; PTX, pertussis toxin; Rac1, Ras-related C3 botulinum toxin substrate 1; ROCK, Rho-associated protein kinase.
FABP7
Fatty acid binding protein 7 (FABP7), a member of the multi-gene FABP family comprised of structurally related proteins with expression patterns specific to cell, tissue and development, binds to very long chain polyunsaturated fatty acids (VLCPUFAs) such as docosahexaenoic acid (DHA) with high affinity [422,423]. FABP7 abundant in astrocytes [424-426] is a lipid chaperone mediating cellular uptake, intracellular trafficking and subsequent oxidation of fatty acids (FAs), whose expression was reported to be elevated in GBM and GBM stem-like cells forming neurospheres (NS) and might accounting for GBM aggressiveness [427,428] and recurrence as well as associated with proliferation, migration and invasion of GBM cells, GBM histology and reduced survival time [429-434]. Under metabolic stresses (e.g. hypoxia), fatty acids are stored as lipid droplets (LDs) and subsequently oxidized in a FABP-dependent manner for energy production in GBM cells [435]. Slowcycling cells (SCCs), a subpopulation of GBM cells preferentially utilizing mitochondrial oxidative phosphorylation (OXPHOS), showing elevated lipid contents specifically metabolized under glucose deprivation and displaying enhanced capabilities of migration, invasion and chemoresistance, have been revealed with the characterization of higher FABP expression and larger LD amounts in cultured conditions of normal oxygen levels or nutrients [436]. Additionally, resistance of SCCs against deprived glucose or inhibited glycolysis could be restrained by FA uptake blocking via genetic deletion or pharmacological inhibition of FABP7 [436].
Moreover, promotion effects of FABP7 on GBM cell migration can be mitigated with DHA supplementation through specific and dramatic inhibition of DHA supplementation in culture medium on plasma membrane lipid order of FABP7expressing GBM cells which positively correlates with GBM cell migration as well as DHA supplementation-mediated disruption of nanodomains formed by FABP7 on GBM cell membranes [437], further suggesting a critical role of FABP7 in lipid metabolism in GBM cells.
SCD
Stearoyl-CoA desaturase (SCD) is an endoplasmic reticulum (ER)-localized delta-9 fatty acid desaturase forming carbon-carbon double bonds at the 9th to 10th position from the COOH-terminus of saturated fatty acids (SFAs), stearic acid and palmitic acid and thus generating monounsaturated fatty acids (MUFAs), oleic acid and palmitoleic acid, respectively [438,439], whose expression correlates with the ratio of MUFA to SFA in which a disequilibrium contributes to alterations in cell growth and differentiation [438-441]. SCD has 4 isoforms in mice (SCD1, SCD2, SCD3 and SCD4), while only two paralogs are expressed in human, namely SCD sharing approximately 85% amino acid identity with mouse SCDs and SCD5 unique to primates [440,442]. SCD has been described as a hypermethylated gene member contributing to the CpG island methylator phenotype which defines a distinct glioma subgroup [443]. SCD expression in GBM, in contrast to SCD upregulation often observed in multiple human tumors [444-447], was reported to be lower than normal brain tissues because of hypermethylation and monoallelic deletion together with phosphatase and tensin homolog (PTEN) frequently deleted in GBM [448] in a subset of GBM patients [449]. In addition, GBM cells without epigenetic and genetic changes mentioned above were revealed to express elevated SCD levels on which tumor cells rely for their survival [449]. SCD inhibition by CAY10566, an inhibitor with a modest BBB penetration ability, has been demonstrated to not only significantly suppress intracranial GBM growth, but also obviously affect tumor vasculature including nearly complete blocking of intratumoral bleeding and possible normalization of blood vessels, potentially allowing enhanced delivery of combinedly used antitumor drugs such as temozolomide (TMZ) [449,450].
Conclusions
The brain is highly enriched in lipids where they are crucial for multiple physiological processes ranging from maintenance of structural integrity and metabolic homeostasis to brain function and development. Metabolism of lipids is a complicated process in which a wide range of lipid-related effector proteins are involved and whose alteration is strongly associated with brain dysfunctions and diseases such as Alzheimer’s disease (AD) and glioblastoma (GBM). In this review, we throw light upon basic classes of lipids including fatty acids, phospholipids, sphingolipids, sterol lipids and triglycerides, of which dysregulated metabolism can be regarded as disease biomarkers. We also briefly discuss the role of lipids within the brain and altered lipid profile correlated with astrocytic function and astrocyte-neuron crosstalk in AD and GBM. Moreover, we have discussed lipid-related metabolites and proteins critical for disease-associated lipid dyshomeostasis and how these proteins together with lipids in correlation with astrocytic functions modulate disease pathogenesis and development, enlightening their therapeutic potential in preventing onset and progression of AD and GBM. However, there are still several lipids whose association with AD and GBM and availability as clinically valuable biomarkers for disease detection at early stages need further evaluation, which can be performed by newly-developed and improved techniques of gradually matured lipidomic platforms. What’s more, there remains much to be discovered about benefits and risks of manipulation of compounds affecting effector proteins involved in lipid metabolism, and further characterization of pathways in which important lipid-related proteins participate along with clinical studies will aid the understanding of pathogenesis mechanisms behind AD and GBM and identification of novel therapeutic targets to help ameliorate disease courses, facilitate disease treatments and consequently benefit patients.
References
- Mita R, Coles JE, Glubrecht DD, Sung R, Sun X, et al. (2007) B-FABP-expressing radial glial cells: The malignant glioma cell of origin? Neoplasia 9: 734-744. [crossref]
- Mita R, Beaulieu MJ, Field C, Godbout R (2010) Brain fatty acid-binding protein and omega-3/omega-6 fatty acids: Mechanistic insight into malignant glioma cell migration. J Biol Chem 285: 37005-37015. [crossref]
- Antonella DR, Serena P, Marco R, Patrizia T, Letizia M, et al. (2012) A radial glia gene marker, fatty acid binding protein 7 (FABP7), is involved in proliferation and invasion of glioblastoma cells. Plos One 7: e52113. [crossref]
- (2020) 2020 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia 16: 391-460.
- Brookmeyer R, Evans DA, Hebert L, Langa KM, Heeringa SG, et al. (2011) National estimates of the prevalence of Alzheimer’s disease in the United States. Alzheimer’s & Dementia 7: 61-73. [crossref]
- Rlhh A, Cagw A, Pa A, Maj B, Frjv A, Jls C (2014) Diagnosing Alzheimer’s disease: A systematic review of economic evaluations. Alzheimer’s & Dementia 10: 225-237.
- Wilfling F, Haas JT, Walther TC, Jr R (2014) Lipid droplet biogenesis. Curr Opin Cell Biol 59: 88-96.
- Olzmann James A, Carvalho P (2019) Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol 20: 137-155. [crossref]
- Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, et al. (2015) Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 160: 177-190.
- Yoon H, Shaw JL, Haigis MC, Greka A (2021) Lipid metabolism in sickness and in health: Emerging regulators of lipotoxicity. Mol Cell 81: 3708-3730. [crossref]
- Ioannou MS, Jackson J, Sheu SH, Chang CL, Liu Z (2019) Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 177: 1522-1535.
- Liu L, Mackenzie KR, Putluri N, Maleti-Savati M, Bellen HJ (2017) The Glia-Neuron lactate shuttle and elevated ROS promote lipid synthesis in neurons and lipid droplet accumulation in glia via APOE/D. Cell Metab 26: 719-737. [crossref]
- Magistretti PJ, Allaman I (2018) Lactate in the brain: From metabolic endproduct to signalling molecule. Nat Rev Neurosci 19: 235-249. [crossref]
- Magistretti PPJ (1994) Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proceedings of the National Academy of ences of the United States of America 91: 10625-10629. [crossref]
- Alexei, Verkhratsky, Vladimir, Parpura, Nina, Vardjan, et al. (2019) Physiology of astroglia. Adv Exp Med Biol 1175: 45-91.
- Afridi R, Kim JH, Rahman MH, Suk K (2020) Metabolic regulation of glial phenotypes: Implications in neuron-glia interactions and neurological disorders. Front Cell Neurosci 14: 20. [crossref]
- Sharma K, Schmitt S, Bergner CG, Tyanova S, Kannaiyan N, et al. (2015) Cell type- and brain region-resolved mouse brain proteome. Nat Neurosci 18: 1819-1831. [crossref]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O Keeffe S, et al. (2014) An RNA-Sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34: 11929-11947. [crossref]
- Abe T, Takahashi S, Suzuki N (2006) Oxidative metabolism in cultured rat astroglia: Effects of reducing the glucose concentration in the culture medium and of D-aspartate or potassium stimulation. Journal of Cerebral Blood Flow & Metabolism J Cereb Blood Flow Metab 26: 153-160. [crossref]
- Goyal MS, Hawrylycz M, Miller JA, Snyder AZ, Raichle ME (2014) Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metab 19: 49-57. [crossref]
- Dienel GA (2019) Brain glucose metabolism: Integration of energetics with function. Physiol Rev 99: 949-1045. [crossref]
- Dienel GA, Cruz NF (2016) Aerobic glycolysis during brain activation: Adrenergic regulation and influence of norepinephrine on astrocytic metabolism. J Neurochem 138: 14-52. [crossref]
- Ramasamy Indra (2014) Recent advances in physiological lipoprotein metabolism. Clin Chem Lab Med 52. [crossref]
- Filou S, Lhomme M, Karavia EA, Kalogeropoulou C, Theodoropoulos V, et al (2016) Distinct roles of apolipoproteins a1 and e in the modulation of High-Density lipoprotein composition and function. Biochemistry 55: 3752-3762 [crossref]
- Bolanos-Garcia VM, Miguel RN (2003) On the structure and function of apolipoproteins: More than a family of lipid-binding proteins. Prog Biophys Mol Biol 83: 47-68. [crossref]
- Ito JI, Nagayasu Y, Miura Y, Yokoyama S, Michikawa M (2014) Astrocytes endogenous apoE generates HDL-like lipoproteins using previously synthesized cholesterol through interaction with ABCA1. Brain Res 1570: 1-12. [crossref]
- Center M, Francisco S Introduction to lipids and lipoproteins.
- Pitas RE, Boyles JK, Lee SH, Foss D, Mahley RW (1987) Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim Biophys Acta 917: 148-161. [crossref]
- Balazs Z, Panzenboeck U, Hammer A, Sovic A, Quehenberger O, et al. (2010) Uptake and transport of high-density lipoprotein (HDL) and HDLassociated alpha-tocopherol by an in vitro blood-brain barrier model. J Neurochem 89: 939-950. [crossref]
- Braun V, Hantke K (2019) Lipoproteins: Structure, function, biosynthesis. Subcell Biochem 92: 39-77. [crossref]
- Hoofnagle AN, Heinecke JW (2009) Lipoproteomics: Using mass spectrometry-based proteomics to explore the assembly, structure, and function of lipoproteins. J Lipid Res 50: 1967-1975. [crossref]
- Alaupovic P (1996) Significance of apolipoproteins for structure, function, and classification of plasma lipoproteins. Method Enzymol 263: 32-60. [crossref]
- Ghosh S, Strum JC, Bell RM (1997) Lipid biochemistry: Functions of glycerolipids and sphingolipids in cellular signaling. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 11: 45. [crossref]
- Zhu X, Owen JS, Wilson MD, Li H, Griffiths GL, et al. (2010) Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J Lipid Res 51: 3196-3206. [crossref]
- Woller SA, Choi S, An EJ, Low H, Schneider DA, Ramachandran R, et al. (2018) Inhibition of neuroinflammation by AIBP: Spinal effects upon facilitated pain states. Cell Rep 23: 2667-2677. [crossref]
- Zhang J, Li Q, Wu Y, Wang D, Xu L, Zhang Y, et al. (2019) Cholesterol content in cell membrane maintains surface levels of ErbB2 and confers a therapeutic vulnerability in ErbB2-positive breast cancer. Cell Commun Signal 17. [crossref]
- Banach-Orowska M, Wyszyska R, Pyrzyska B, Maksymowicz M, et al. (2019) Cholesterol restricts lymphotoxin receptor-triggered NF-B signaling. Cell Commun Signal 17. [crossref]
- Luchtman DW, Song C (2013) Cognitive enhancement by omega-3 fatty acids from child-hood to old age: Findings from animal and clinical studies. Neuropharmacology 64: 550-565. [crossref]
- Hamilton JA, Hillard CJ, Spector AA, Watkins PA (2007) Brain uptake and utilization of fatty acids, lipids and lipoproteins: Application to neurological disorders. J Mol Neurosci 33: 2-11. [crossref]
- Dawson G 1999 Basic Neurochemistry, 6th Edition. J Neurosci Res
- Subramaniam S, Fahy E, Gupta S, Sud M, Byrnes RW, et al. (2011) Bioinformatics and systems biology of the lipidome. Chem Rev 111: 6452-6490. [crossref]
- Han X (2016) Lipidomics for studying metabolism. Nat Rev Endocrinol 12: 668-679. [crossref]
- Li M, Yang L, Bai Y, Liu H (2014) Analytical methods in lipidomics and their applications. Anal Chem 86: 161-175. [crossref]
- Cermenati G, Mitro N, Audano M, et al. (2015) Lipids in the nervous system: From biochemistry and molecular biology to patho-physiology. Biochim Biophys Acta 1851: 51-60. [crossref]
- Qiuhui X, Chunxiu H, Di Y, Melcangi RC, Crestani M, et al. (2018) Development of a high coverage pseudotargeted lipidomics method based on UltraHigh performance liquid Chromatography-Mass spectrometry. Anal Chem 90: 7608-7616. [crossref]
- Wood PL, Cebak JE (2018) Lipidomics biomarker studies: Errors, limitations, and the future. Biochem Biophys Res Commun 504: 569-575. [crossref]
- Rustam YH, Reid GE (2017) Analytical challenges and recent advances in mass spectrometry based lipidomics. Anal Chem 90: 374-397. [crossref]
- Chaves ED, Rusinol AE, Vance DE, Campenot RB, Vance JE (1997) Role of lipoproteins in the delivery of lipids to axons during axonal regeneration. J Biol Chem 272: 30766-30773. [crossref]
- Fester L, Zhou L, Bütow A, Huber C, von Lossow R, et al. (2009) Cholesterol promoted synaptogenesis requires the conversion of cholesterol to estradiol in the hippocampus. Hippocampus 19: 692-705. [crossref]
- Sang N, Chen C (2006) Lipid signalling and synaptic plasticity. Neuroscientist 12: 425-434. [crossref]
- Hussain G, Wang J, Rasul A, et al. (2019) Role of cholesterol and sphingolipids in brain development and neurological diseases. Lipids Health Dis 18: 1-2. [crossref]
- Jorge C, Farez MF (2015) The role of astrocytes in multiple sclerosis progression. Front Neurol 6: 180. [crossref]
- Joe EH, Choi DJ, An J, et al. (2018) Astrocytes, microglia, and parkinson’s disease. Exp Neurobiol 27: 77-87. [crossref]
- Almad A, Maragakis NJ (2018) A stocked toolbox for understanding the role of astrocytes in disease. Nat Rev Neurol 14: 351-362. [crossref]
- Belanger M, Allaman I, Magistretti PJ (2011) Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab 14: 724-738. [crossref]
- Ullian EM, Sapperstein SK, Christopherson KS, et al. (2001) Control of synapse number by glia. Science 291: 657-661. [crossref]
- Varcianna A, Myszczynska MA, Castelli LM, et al. (2018) Micro-RNAs secreted through astrocyte-derived extracellular vesicles cause neuronal network degeneration in C9orf72 ALS. Ebiomedicine 40: 626-635. [crossref]
- Chung WS, Allen NJ, Eroglu C (2015) Astrocytes control synapse formation, function, and elimination. Cold Spring Harb Perspect Biol 7: a020370. [crossref]
- Sofroniew MV, Vinters HV, Vinters HV (2013) REVIEW Astrocytes: Biology and pathology. Acta Neuropathol 119: 7-35. [crossref]
- Deitmer JW, Theparambil SM, Ruminot I, et al. (2019) Energy dynamics in the brain: Contributions of astrocytes to metabolism and pH homeostasis. Front Neurosci-Switz 13: 1301. [crossref]
- Ballabh P, Braun A, Nedergaard M (2004) The blood-brain barrier: An overview: Structure, regulation, and clinical implications. Neurobiol Dis 16: 1-13. [crossref]
- Abbott NJ, Patabendige AAK, Dolman DEM, et al (2010) Structure and function of the blood–brain barrier. Neurobiol Dis 37: 13-25. [crossref]
- Edmond J (2001) Essential polyunsaturated fatty acids and the barrier to the brain. J Mol Neurosci 16: 181-193. [crossref]
- Mitchell RW, Hatch GM (2011) Fatty acid transport into the brain: Of fatty acid fables and lipid tails. Prostaglandins Leukot Essent. Fatty Acids 85: 293-302. [crossref]
- Saunders NR, Ek CJ, Habgood MD (2008) Barriers in the brain: A renaissance? Trends Neurosci 31: 279-286. [crossref]
- Bradbury MW (1984) The structure and function of the blood-brain barrier. Federation Proceedings 43: 186-190.
- Betsholtz C (2014) Physiology: Double function at the blood brain barrier. Nature 509: 432-433.
- Daneman R, Prat A (2015) The Blood鈥揃rain Barrier. Csh Perspect Biol 7: 32-34.
- Ferreira L (2019) What human blood-brain barrier models can tell us about BBB function and drug discovery? Expert Opin Drug Dis 14: 1113-1123. [crossref]
- Moura RP, Martins C, Pinto S, et al. (2019) Blood-brain barrier receptors and transporters: An insight on their function and how to exploit them through nanotechnology. Expert Opin Drug Del 16: 271-285. [crossref]
- Dienel GA, Cruz NF (2016) Aerobic glycolysis during brain activation: Adrenergic regulation and influence of norepinephrine on astrocytic metabolism. J Neurochem 138: 14-52. [crossref]
- Goyal MS, Hawrylycz M, Miller JA, et al. (2014) Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metab 19: 49-57. [crossref]
- Abe T, Takahashi S, Suzuki N (2006) Oxidative metabolism in cultured rat astroglia: Effects of reducing the glucose concentration in the culture medium and of D-aspartate or potassium stimulation. J Cereb Blood Flow Metab 26: 153-160. [crossref]
- Cataldo AM, Broadwell RD (1986) Cytochemical identification of cerebral glycogen and glucose-6-phosphatase activity under normal and experimental conditions. II. Choroid plexus and ependymal epithelia, endothelia and pericytes. J Neurocytol 15: 511-524. [crossref]
- Pfeiffer-Guglielmi B, Fleckenstein B, Jung G, et al. (2010) Immunocytochemical localization of glycogen phosphorylase isozymes in rat nervous tissues by using isozyme-specific antibodies. J Neurochem 85: 73-81. [crossref]
- Camargo N, Goudriaan A, Deijk ALFV, et al. (2017) Oligodendroglial myelination requires astrocyte-derived lipids. Plos Biol 15: e1002605. [crossref]
- Van Deijk ALF, Camargo N, Timmerman J, et al. (2017) Astrocyte lipid metabolism is critical for synapse development and function in vivo. 65: 670-682. [crossref]
- Ioannou MS, Jesse J, Shu-Hsien S, et al. (2020) Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 177: 1522-1535. [crossref]
- Ebert D, Haller RG, Walton ME (2003) Energy contribution of octanoate to intact rat brain metabolism measured by 13C nuclear magnetic resonance spectroscopy. J Neurosci 23: 5928-5935. [crossref]
- Chang CY, Ke DS, Chen JY (2009) Essential fatty acids and human brain. Acta Neurol Taiwan 18: 231-241. [crossref]
- Edmond J, Robbins RA, Bergstrom JD, et al. (2010) Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J Neurosci Res 18: 551-561. [crossref]
- Takahashi S, Iizumi T, Mashima K, et al. (2014) Roles and regulation of ketogenesis in cultured astroglia and neurons under hypoxia and hypoglycemia. Asn Neuro 6: [crossref]
- Simard JR, Pillai BK, Hamilton JA (2008) Fatty acid flip-flop in a model membrane is faster than desorption into the aqueous phase. Biochemistry 47: 9081-9089. [crossref]
- Mitchell RW, On NH, Bigio MRD, et al. (2011) Fatty acid transport protein expression in human brain and potential role in fatty acid transport across human brain microvessel endothelial cells. J Neurochem 117: 735-746. [crossref]
- Marianne Haag (2003) Essential fatty acids and the brain. Can J Psychiatry 48: 195-203.
- Tracey TJ, Steyn FJ, Wolvetang EJ, et al. (2018) Neuronal lipid metabolism: Multiple pathways driving functional outcomes in health and disease. Front Mol Neurosci 11: 10. [crossref]
- Crawford MA, Casperd NM, Sinclair AJ (1976) The long chain metabolites of linoleic and linolenic acid in liver and brain in herbivores and carnivores. Comparative biochemistry and physiology. Comp Biochem Physiol B 54: 395-401. [crossref]
- Crawford MA, Broadhurst CL, Guest M, et al. (2013) A quantum theory for the irreplaceable role of docosahexaenoic acid in neural cell signalling throughout evolution. Prostaglandins Leukot Essent Fatty Acids 88: 5-13. [crossref]
- Bazinet RP, Lay S (2014) Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat Rev Neurosci 15: 771-785. [crossref]
- Luchtman DW, Song C (2013) Cognitive enhancement by omega-3 fatty acids from child-hood to old age: Findings from animal and clinical studies. 64: 550-565. [crossref]
- Denis I, Potier B, Vancassel S, et al (2013) Omega-3 fatty acids and brain resistance to ageing and stress: Body of evidence and possible mechanisms. Ageing Res Rev 12: 579-594. [crossref]
- Igarashi M, Santos RA, Cohen-Cory S (2015) Impact of Maternal n-3 Polyunsaturated Fatty Acid Deficiency on Dendritic Arbor Morphology and Connectivity of DevelopingXenopus laevisCentral NeuronsIn Vivo. J Neurosci 35: 6079-6092. [crossref]
- Lim GP, Calon F, Morihara T, et al. (2005) A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged alzheimer mouse model. J Neurosci 25: 3032-3040. [crossref]
- Michael A, Crawford (2006) Docosahexaenoic acid in neural signaling systems. Nutrition & Health
- Hamilton JA, Hillard CJ, Spector AA, et al. (2007) Brain uptake and utilization of fatty acids, lipids and lipoproteins: Application to neurological disorders. J Mol Neurosci 33: 2-11. [crossref]
- Farooqui AA, Horrocks LA, Farooqui T (2000) Glycerophospholipids in brain: Their metabolism, incorporation into membranes, functions, and involvement in neurological disorders. Chem Phys Lipids 106: 1-29. [crossref]
- Van Meer G, Voelker DR, Feigenson GW (2008) Membrane lipids: Where they are and how they behave. Nat Rev Mol Cell Bio 9: 112-124. [crossref]
- Harayama T, Riezman H (2018) Understanding the diversity of membrane lipid composition. Nat Rev Mol Cell Biol 19: 281-296. [crossref]
- Li J, Wang X, Zhang T, et al. (2015) A review on phospholipids and their main applications in drug delivery systems. Asian J Pharm Sci 10: 81-98. [crossref]
- Ross BM, Moszczynska A, Blusztajn JK, et al. (1997) Phospholipid biosynthetic enzymes in human brain. Lipids 32: 351-358. [crossref]
- Paltauf F, Hermetter A (1990) Phospholipids natural, semisynthetic, synthetic: Phospholipids 1-12. [crossref]
- Tracey TJ, Steyn FJ, Wolvetang EJ, et al. (2018) Neuronal lipid metabolism: Multiple pathways driving functional outcomes in health and disease. Front Mol Neurosci 11: 10. [crossref]
- Montaner A, da Silva Santana TT, Schroeder T, et al. (2018) Author correction: Specific phospholipids regulate the acquisition of neuronal and astroglial identities in Post-Mitotic cells. Sci Rep 9: 20222. [crossref]
- Sonnino S, Prinetti A (2012) Membrane domains and the lipid raft concept. Curr Med Chem. 20: 4-21. [crossref]
- West RJ, Briggs L, Perona Fjeldstad M (2018) Sphingolipids Regulate Neuromuscular Synapse Structure and function in Drosophila. J Comp Neurol 526: 1995-2009. [crossref]
- Vance Dennis E (2008) Biochemistry of lipids, lipoproteins and membranes (Fifth edition).:v-vi
- Hannun YA, Obeid LM (2018) Author Correction: Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Bio 19: 673. [crossref]
- Schnaar Ronald L (2016) Gangliosides of the vertebrate nervous system. J Mol Biol 428: 3325-3336. [crossref]
- Novgorodov SA, Voltin JR, Wang W, et al. (2019) Acid sphingomyelinase deficiency protects mitochondria and improves function recovery after brain injury. J Lipid Res 60: 609-623. [crossref]
- Hartmann D, Lucks J, Fuchs S, et al. (2012) Long chain ceramides and very long chain ceramides have opposite effects on human breast and colon cancer cell growth. Int J Biochem Cell Biol 44: 620-628. [crossref]
- Xu R, Wang K, Mileva I, et al. (2016) Alkaline ceramidase 2 and its bioactive product sphingosine are novel regulators of the DNA damage response. Oncotarget 7: 18440-18457. [crossref]
- Gomez-Mu Oz A, Presa N, Gomez-Larrauri A, et al. (2016) Control of inflammatory responses by ceramide, sphingosine 1phosphate and ceramide 1-phosphate. Prog Lipid Res 61: 51-62. [crossref]
- Blom T, Somerharju P, Ikonen E (2011) Synthesis and biosynthetic trafficking of membrane lipids. Cold Spring Harb Perspect Biol 3: a4713. [crossref]
- Björkhem I, Meaney S (2004) Brain cholesterol: Long secret life behind a barrier. Arterioscler Thromb Vasc Biol 24: 806-815. [crossref]
- Kang S, Kim CH, Jung H, et al. (2017) Agmatine ameliorates type 2 diabetes induced-Alzheimer’s disease-like alterations in high-fat diet-fed mice via reactivation of blunted insulin signalling. Neuropharmacology 113: 467-479. [crossref]
- Saeed AA, Genove G, Li T, Lutjohann D, Olin M, et al. (2014) Effects of a disrupted Blood-Brain barrier on cholesterol homeostasis in the brain. J Biol Chem [crossref]
- Reinhart MP, Billheimer JT, Faust JR, Gaylor JL (1987) Subcellular localization of the enzymes of cholesterol biosynthesis and metabolism in rat liver. J Biol Chem 262: 9649. [crossref]
- Muse ED, Jurevics H, Toews AD, Matsushima GK, Morell P (2010) Parameters related to lipid metabolism as markers of myelination in mouse brain. J Neurochem 76: 77-86. [crossref]
- Zarrouk, Amira, Debbabi, Meryam, Bezine, Maryem, et al. (2018) Lipid biomarkers in alzheimer’s disease. Curr Alzheimer Res 15: 303-312. [crossref]
- Genaro-Mattos TC, Anderson A, Allen LB, Korade Z, Mirnics K (2019) Cholesterol biosynthesis and uptake in developing neurons. Acs Chem Neurosci [crossref]
- Deijk V, Anne-Lieke F, Camargo, Nutabi, Timmerman Jaap, et al. (2017) Astrocyte lipid metabolism is critical for synapse development and function in vivo. Glia 65: 670-682. [crossref]
- Wang H, Eckel RH (2014) What are lipoproteins doing in the brain? Trends in Endocrinology & Metabolism Tem 25: 8-14. [crossref]
- Orth M, Bellosta S (2012) Cholesterol: Its regulation and role in central nervous system disorders. Cholesterol 2012: 292598. [crossref]
- Pitas RE, Boyles JK, Lee SH, Hui D, Weisgraber KH (1987) Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E (LDL) receptors in the brain. J Biol Chem 262: 14352-14360. [crossref]
- Jing C, Zhang X, Kusumo H, Costa LG, Guizzetti M (2013) Cholesterol efflux is differentially regulated in neurons and astrocytes: Implications for brain cholesterol homeostasis. Biochim Biophys Acta 1831. [crossref]
- Rushworth JV, Hooper NM (2010) Lipid rafts: Linking alzheimer’s amyloidbeta production, aggregation, and toxicity at neuronal membranes. International Journal of Alzheimer’s Disease 2011: 603052. [crossref]
- Kabouridis PS, Janzen J, Magee AL, Ley SC (2015) Cholesterol depletion disrupts lipid rafts and modulates the activity of multiple signaling pathways in T lymphocytes. Eur J Immunol 30: 954-963. [crossref]
- Pike LJ (2003) Lipid rafts bringing order to chaos. J Lipid Res 44: 655-667. [crossref]
- Hussain G, Wang J, Rasul A, Anwar H, Imran A, et al. (2019) Role of cholesterol and sphingolipids in brain development and neurological diseases. Lipids Health Dis 18. [crossref]
- Mauch DH, Ngler K, Schumacher S, Gritz C, Miller E, Otto A, et al. (2001) CNS synaptogenesis promoted by Glia-Derived cholesterol. Science 294. [crossref]
- Fukui K, Ferris HA, Kahn CR (2015) Effect of Cholesterol Reduction on Receptor Signaling in Neurons. J Biol Chem 290: 26383-26392. [crossref]
- Chaves ED, Rusinol AE, Vance DE, Campenot RB, Vance JE (1997) Role of lipoproteins in the delivery of lipids to axons during axonal regeneration. J Biol Chem 272: 30766. [crossref]
- Xue-Shan Z, Juan P, Qi W, Zhong R, Li-Hong P, et al. (2016) Imbalanced cholesterol metabolism in Alzheimer’s disease. Clin Chim Acta 456: 107-114. [crossref]
- Kandimalla R, Thirumala V, Reddy PH (2016) Is Alzheimer’s disease a Type 3 Diabetes? A Critical Appraisal. Biochimica et Biophysica Acta 1863: 1078-1089. [crossref]
- S Sonnino, A Prinetti (2013) Membrane domains and the “lipid raft” concept. Curr Med Chem [crossref]
- Caliceti C, Zambonin L, Prata C, Sega F, Hakim G, et al. (2012) Effect of plasma membrane cholesterol depletion on glucose transport regulation in leukemia cells. Plos One 7. [crossref]
- Group BMJP (2009) Alzheimer’s disease. BMJ 338.
- Mattson MP (2016) Pathways towards and away from Alzheimer’s disease. Nature 430: 631-639. [crossref]
- Vishal S, Sourabh A, Harkirat S (2011) Alois Alzheimer (1864-1915) and the Alzheimer syndrome. Journal of Medical Biography 19: 32-33. [crossref]
- D’Errico P, Luehmann MM (2020) Mechanisms of pathogenic tau and a protein spreading in alzheimer’s disease. Front Aging Neurosci 12: 265. [crossref]
- Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: 239-259. [crossref]
- El Kadmiri, Said N, Slassi Ilham, Moutawakil BE, Nadifi S (2018) Biomarkers for alzheimer disease: Classical and novel candidates’ review. Neuroscience 370: 181-190. [crossref]
- Foley P (2010) Lipids in Alzheimer’s disease: A century-old story. BBA – Molecular and Cell Biology of Lipids 1801: 750-753. [crossref]
- Alzheimer A (1907) Uber eine eigenartige Erkrankung der Hirnride. Allg.z.psychiatr
- Kim M, Nevado-Holgado A, Whiley L, Snowden SG, Soininen H, et al. (2016) Association between plasma ceramides and phosphatidylcholines and hippocampal brain volume in late onset alzheimer’s disease. Journal of Alzheimers Disease 60: 809-817. [crossref]
- González-Domínguez R, García-Barrera T, Vitorica J, Gómez-Ariza JL (2015) Metabolomic screening of regional brain alterations in the APP/PS1 transgenic model of Alzheimer’s disease by direct infusion mass spectrometry. J Pharmaceut Biomed 102: 425-435. [crossref]
- Bennett S, Valenzuela N, Xu H, Franko B, Fai S, et al. (2013) Using neurolipidomics to identify phospholipid mediators of synaptic (dys)function in Alzheimer’s Disease. Front Physiol 4: 168. [crossref]
- Kao YC, Ho PC, Tu YK, Jou IM, Tsai KJ (2020) Lipids and alzheimer’s disease. Int J Mol Sci 21: 1505. [crossref]
- Grimm MO, Michaelson D, Hartmann T (2017) Omega-3 fatty acids, lipids and apoE lipidation in Alzheimer’s disease: A rationale for multi-nutrient dementia prevention. J Lipid Res 58: 2083-2101. [crossref]
- Wood PL (2012) Lipidomics of Alzheimer’s disease: Current status. Alzheimers Research & Therapy 4: 5. [crossref]
- Bales KR (2010) Brain lipid metabolism, apolipoprotein E and the pathophysiology of Alzheimer’s disease. Neuropharmacology 59: 295-302. [crossref]
- Ledesma MD, Martin MG, Dotti CG (2012) Lipid changes in the aged brain: Effect on synaptic function and neuronal survival. Prog Lipid Res 51: 23-35. [crossref]
- Sultana R, Perluigi M, Allan Butterfield D (2013) Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer disease brain. Free Radical Biology & Medicine 62: 157-169. [crossref]
- Tian-Bi, Zhu, Zhao, Zhang, Piao, Luo, et al. (2018) Lipid metabolism in Alzheimer’s disease. Brain Res Bull
- Nasaruddin ML, Holscher C, Kehoe P, Graham SF, Green BD (2016) Wideranging alterations in the brain fatty acid complement of subjects with late Alzheimer’s disease as detected by GC-MS. Am J Transl Res 8: 154-165. [crossref]
- Nasaruddin ML, Pan X, Mcguinness B, Passmore P, Kehoe PG, et al. (2018) Evidence that parietal lobe fatty acids may be more profoundly affected in moderate alzheimer’s disease (AD) pathology than in severe AD pathology. Metabolites 8: 69. [crossref]
- Wood PL, Barnette BL, Kaye JA, Quinn JF, Woltjer RL (2015) Non-targeted lipidomics of CSF and frontal cortex grey and white matter in control, mild cognitive impairment, and Alzheimer’s disease subjects. Acta Neuropsychiatr 27: 270-278. [crossref]
- Belkouch M, Hachem M, Elgot A, Van AL, Picq M, et al. (2016) The pleiotropic effects of omega-3 docosahexaenoic acid on the hallmarks of Alzheimer’s disease. J Nutr Biochem 38: 1-11. [crossref]
- Sarrafpour S, Ormseth C, Chiang A, Arakaki X, Harrington M, et al. (2019) Lipid Metabolism in Late-Onset Alzheimer’s Disease Differs from Patients Presenting with Other Dementia Phenotypes. Int J Env Res Pub He 16: 1995. [crossref]
- Wong MW, Braidy N, Poljak A, Pickford R, Thambisetty M (2017) Dysregulation of lipids in Alzheimer’s disease and their role as potential biomarkers. Alzheimer’s & Dementia 13: 810-827. [crossref]
- Xiong H, Callaghan D, Jones A, Walker DG, Lue LF, et al. (2008) Cholesterol retention in Alzheimer’s brain is responsible for high – and secretase activities and a production. Neurobiol Dis 29: 422-437. [crossref]
- Pfrieger FW (2003) Cholesterol homeostasis and function in neurons of the central nervous system. Cellular & Molecular Life Sciences 60: 1158-1171. [crossref]
- Chang TY, Yamauchi Y, Hasan MT, Chang C (2017) Cellular cholesterol homeostasis and Alzheimers J Lipid Res 58: 2239-2254. [crossref]
- Scala CD, Chahinian H, Yahi N, Garmy N, Fantini J (2014) Interaction of alzheimer’s-Amyloid peptides with cholesterol: Mechanistic insights into amyloid pore formation. Biochemistry 53: 4489-4502. [crossref]
- Area-Gomez E, Maria DCLC, Tambini MD, Guardia-Laguarta C, De Groof AJC, et al. (2012) Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. Embo J 31: 4106-4123.
- Area-Gomez E, Schon EA (2016) Mitochondria-associated ER membranes and Alzheimer disease. Curr Opin Genet Dev 38: 90-96. [crossref]
- Pera M, Larrea D, Guardia Laguarta C, Montesinos J, Velasco KR, Agrawal RR, et al. (2017) Increased localization of APP C99 in mitochondria associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. The EMBO Journal 36: 3356-3371. [crossref]
- Pera M, Montesinos J, Larrea D, Agrawal RR, Area-Gomez E (2020) MAM and C99, key players in the pathogenesis of Alzheimer’s disease. Int Rev Neurobiol 154: 235-278. [crossref]
- Andrew P Bailey, Grielof Koster, Christelle Guillermier, Hirst EMA, Macrae JI, et al. (2015) Antioxidant role for lipid droplets in a stem cell niche of drosophila. Cell 163: 340-353. [crossref]
- Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, et al. (2015) Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 160: 177-190. [crossref]
- Liu L, Mackenzie KR, Putluri N, Maleti-Savati M, Bellen HJ (2017) The Glia-Neuron lactate shuttle and elevated ROS promote lipid synthesis in neurons and lipid droplet accumulation in glia via APOE/D. Cell Metab 26: 719-737. [crossref]
- Wong MW, Braidy N, Poljak A, Sachdev PS (2017) The application of lipidomics to biomarker research and pathomechanisms in Alzheimer’s disease. Curr Opin Psychiatr 30: 136-144. [crossref]
- Zhamg AH, Ma MZ, Kong L, Gao HL, Sun H (2020) High-throughput lipidomics analysis to discover lipid biomarkers and profiles as potential targets for evaluating efficacy of an against APP/PS1 transgenic mice based on UPLC S. Biomed Chromatogr 34: 4724. [crossref]
- Barupal DK, Baillie R, Fan S, Saykin AJ, Kaddurah-Daouk R (2019) Sets of coregulated serum lipids are associated with Alzheimer’s disease pathophysiology. Alzheimer s & Dementia Diagnosis Assessment & Disease Monitoring 11: 619-627. [crossref]
- Kim J, Basak JM, Holtzman DM (2009) The role of apolipoprotein e in alzheimer’s disease. Neuron 63: 287-303. [crossref]
- Monteiro-Cardoso VF, Oliveira MM, Melo T, Domingues MRM, Videira RA (2015) Cardiolipin profile changes are associated to the early synaptic mitochondrial dysfunction in alzheimer’s disease. Journal of Alzheimers Disease 43: 1375-1392. [crossref]
- Basu Ball W, Neff JK, Gohil VM (2017) The role of nonbilayer phospholipids in mitochondrial structure and function. Febs Lett 592: 1273-1290. [crossref]
- Calzada E, Onguka O, Claypool SM (2016) Phosphatidylethanolamine metabolism in health and disease. Int Rev Cel Mol Bio 321: 29-88. [crossref]
- Fonteh AN, Cipolla M, Chiang J, Arakaki X, Harrington MG (2014) Human cerebrospinal fluid fatty acid levels differ between supernatant fluid and Brain Derived nanoparticle fractions, and are altered in alzheimer’s disease. Plos One 9: 100519. [crossref]
- Giudetti AM, Shu G, Vergara D, Fonteh AN (2020) Polyunsaturated fatty acid composition of cerebrospinal fluid fractions shows their contribution to cognitive resilience of a pre-symptomatic alzheimer’s disease cohort. Front Physiol 11: 1-14.
- Wilde MD, Vellas B, Girault E, Yavuz AC, Sijben JW (2017) Lower brain and blood nutrient status in Alzheimer’s disease: Results from meta-analyses. Alzheimers & Dementia: Translational Research & Clinical Interventions 3: 416-431. [crossref]
- Snowden SG, Ebshiana AA, Hye A, An Y, Thambisetty M (2017) Association between fatty acid metabolism in the brain and Alzheimer disease neuropathology and cognitive performance: A nontargeted metabolomic study. Plos Med 14: 1002266. [crossref]
- Mh A, Apab C, Nb A, Jc A, Psa D (2020) Blood fatty acids in Alzheimer’s disease and mild cognitive impairment: A meta-analysis and systematic review. Ageing Res Rev 60: 101043. [crossref]
- Wood PL, Barnette BL, Kaye JA, Quinn JF, Woltjer RL (2015) Non-targeted lipidomics of CSF and frontal cortex grey and white matter in control, mild cognitive impairment and Alzheimer’s disease subjects. Acta Neuropsychiatr 27: 270-278. [crossref]
- Dyall SC (2015) Long-chain omega-3 fatty acids and the brain: A review of the independent and shared effects of EPA, DPA and DHA. Front Aging Neurosci 7: 52. [crossref]
- Di Iorio, Benedetta Bartali, Annamaria Corsi, Stefania Bandinelli, Mark P Mattson, et al. (2007) Low plasma N-3 fatty acids and dementia in older persons: The InCHIANTI study. J Gerontol A Biol Med 62: 1120-1126. [crossref]
- Thomas MH, Pelleieux S, Vitale N, Olivier JL (2016) Dietary arachidonic acid as a risk factor for age-associated neurodegenerative diseases: Potential mechanisms. Biochimie: 168-177. [crossref]
- Goozee K, Chatterjee P, James I, Shen K, Sohrabi HR, Asih PR, et al. (2017) Alterations in erythrocyte fatty acid composition in preclinical Alzheimer’s disease. Sci Rep-Uk
7. - Prasad MR, Lovell MA, Yatin M, Dhillon H, Markesbery WR (1998) Regional membrane phospholipid alterations in alzheimer’s disease. Neurochem Res 23: 81-88. [crossref]
- Cunnane SC, Schneider JA, Tangney C, Tremblay-Mercier J, Morris MC (2012) Plasma and brain fatty acid profiles in mild cognitive impairment and alzheimer’s disease. Journal of Alzheimer’s disease: JAD 29: 691-697. [crossref]
- Iwamoto N, Kobayashi K, Kosaka K (1989) The formation of prostaglandins in the postmortem cerebral cortex of Alzheimer-type dementia patients. J Neurol 236: 80-84. [crossref]
- Wood PL (2012) Lipidomics of Alzheimer’s disease: Current status. Alzheimers Research & Therapy 4: 5. [crossref]
- Fonteh AN, Chiang J, Cipolla M, Hale J, Diallo F, et al. (2013) Alterations in cerebrospinal fluid glycerophospholipids and phospholipase A2 activity in Alzheimer’s disease. J Lipid Res 54: 2884-2897. [crossref]
- A LW, A AS, B JH, A PP, C DG, et al. (2014) Evidence of altered phosphatidylcholine metabolism in Alzheimer’s disease. Neurobiol Aging 35: 271-278. [crossref]
- Guan Z, Wang Y, Cairns NJ, Lantos PL, Gustav D, et al. (1999) Decrease and structural modifications of phosphatidylethanolamine plasmalogen in the brain with alzheimer disease. J Neuropathol Exp Neurol 58: 740-747. [crossref]
- Farooqui AA, Horrocks LA (1998) Plasmalogen-selective phospholipase A2 and its involvement in Alzheimer’s disease. Biochem Soc T 26: 243-246. [crossref]
- Filippov V, Song MA, Zhang K, Vinters HV, Duerksen-Hughes PJ (2012) Increased ceramide in brains with alzheimer’s and other neurodegenerative diseases. J Alzheimers Dis 29(3):537-547. [crossref]
- Alfred N Fonteh, Cora Ormseth, Jiarong Chiang, Cipolla M, Arakaki X, et al. (2015) Sphingolipid metabolism correlates with cerebrospinal fluid Beta amyloid levels in Alzheimer’s disease. Plos One 10: 0125597. [crossref]
- Lepara O, Valjevac A, Alajbegovi A, Zairagi A, Nakas-I indi E (2009) Decreased serum lipids in patients with probable Alzheimer’s disease. Bosnian Journal of Basic Medical ences 9: 215-220. [crossref]
- Bernath MM, Bhattacharyya S, Nho K, Barupal DK, Saykin AJ (2018) Serum triglycerides in Alzheimer’s disease: Relation to neuroimaging and CSF biomarkers. Neurology 94: 2088-2098. [crossref]
- Hascalovici JR, Vaya J, Khatib S, Holcroft CA, Schipper HM (2009) Brain sterol dysregulation in sporadic AD and MCI: Relationship to heme oxygenase-1. J Neurochem 110: 1241-1253. [crossref]
- Heverin M, Bogdanovic N, Lutjohann D, Bayer T, Bjrkhem I (2004) Changes in the levels of cerebral and extracerebral sterols in the brain of patients with Alzheimer’s disease. J Lipid Res 45: 186-193. [crossref]
- Tanzi T RE (2018) The genetics of alzheimer disease.
- Guerreiro RJ, Gustafson DR, Hardy J (2012) The genetic architecture of Alzheimer’s disease: Beyond APP, PSENs and APOE. Neurobiol Aging 33: 437-456. [crossref]
- Holtzman DM, Herz J, Bu G (2012) Apolipoprotein e and apolipoprotein e receptors: Normal biology and roles in alzheimer disease. Csh Perspect Med 2: a6312. [crossref]
- RW Mahley (1988) Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science 240: 622-630. [crossref]
- Kim J, Basak JM, Holtzman DM (2009) The role of apolipoprotein e in alzheimer’s disease. Neuron 63: 287-303. [crossref]
- Serrano-Pozo A, Das S, Hyman BT (2021) APOE and Alzheimer’s disease: Advances in genetics, pathophysiology, and therapeutic approaches. The Lancet Neurology 20: 68-80. [crossref]
- Tachibana M, Holm ML, Liu CC, Shinohara M, Kanekiyo T (2019) APOE4mediated amyloid-beta pathology depends on its neuronal receptor LRP1. J Clin Invest 129. [crossref]
- Castellano JM, Kim J, Stewart FR, Jiang H, Demattos RB, et al. (2011) Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med 3: 57r-89r. [crossref]
- Verghese PB, Castellano JM, Garai K, Wang Y, Hong J, Shah A, et al. (2013) ApoE influences amyloid-{beta} (A{beta}) clearance despite minimal apoE/A{beta} association in physiological conditions. Neuroscience 110: E1807-1816. [crossref]
- Basak JM, Verghese PB, Yoon H, Kim J, Holtzman DM (2012) Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of a uptake and degradation by astrocytes. J Biol Chem 287: 13959-13971. [crossref]
- Kanekiyo T, Zhang J, Liu Q, Liu CC, Bu G (2011) Heparan sulphate proteoglycan and the Low-Density lipoprotein Receptor-Related protein 1 constitute major pathways for neuronal amyloid- uptake. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience 31: 1644-1651. [crossref]
- Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R, et al. (2007) Clearance of amyloid-beta by circulating lipoprotein receptors. Nat Med 13: 1029-1031. [crossref]
- Castellano JM, Deane R, Gottesdiener AJ, Verghese PB, Stewart FR, et al. (2012) Low-density lipoprotein receptor overexpression enhances the rate of brain-to-blood clearance in a mouse model of-amyloidosis. P Natl Acad Sci Usa 109: 15502. [crossref]
- Deane ASBV (2008) The role of the cell surface LRP and soluble LRP in blood-brain barrier Abeta clearance in Alzheimer’s disease. Curr Pharm Design 14. [crossref]
- Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, et al. (1997) Lack of apolipoprotein E dramatically reduces amyloid |[beta]|-peptide deposition. Nat Genet 17: 263-264. [crossref]
- Fryer JD (2005) Human apolipoprotein e4 alters the amyloid-? 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J Neurosci 25: 2803-2810. [crossref]
- Holtzman DM (2000) Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences 97: 2892-2897. [crossref]
- Fagan AM, Watson M, Parsadanian M, Bales KR, Holtzman DM (2002) Human and murine ApoE markedly alters a beta metabolism before and after plaque formation in a mouse model of Alzheimer’s disease. Neurobiol Dis 9: 305-318.
- Shadlen MF (1998) Effects of Age and Ethnicity on the Link Between APOE4 and Alzheimer Disease. JAMA [crossref]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Pericak-Vance MA (1993) Gene dose of Apolipoprotein E type 4 allele and the risk of Alzheimers disease in late onset families. Science 8: 41-43. [crossref]
- Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PHS, et al. (1993) Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology (8). [crossref]
- Roses AD (1996) Apolipoprotein E alleles as risk factors in Alzheimer’s disease. Annu Rev Med 47: 387-400. [crossref]
- Mahley RW, Huang Y, Rall SC (1999) Pathogenesis of type III hyperlipoproteinemia (dysbetalipoproteinemia): Questions, quandaries, and paradoxes. J Lipid Res 40: 1933-1949. [crossref]
- Liu CC, Kanekiyo T, Xu H, Bu G (2013) Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat Rev Neurol 9: 106-118. [crossref]
- Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, et al. (1993) Binding of human apolipoprotein E to synthetic amyloid beta peptide: Isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci U S A 90: 8098-8102. [crossref]
- Lefterov I, Wolfe CM, Fitz NF, et al. (2019) APOE2 orchestrated differences in transcriptomic and lipidomic profiles of postmortem AD brain. Alzheimer’s Research and Therapy 11: 1-6. [crossref]
- Juva K, Verkkoniemi A, Viramo P, Polvikoski T, Kainulainen K, et al. (2000) Apolipoprotein e, cognitive function, and dementia in a general population aged 85 years and over. Int Psychogeriatr 12: 379-387. [crossref]
- O’Donoghue MC, Murphy SE, Zamboni G, Nobre AC, Mackay CE (2018) APOE genotype and cognition in healthy individuals at risk of Alzheimer’s disease: A review. Cortex 104: 103-123. [crossref]
- Trachtenberg AJ, Filippini N, Cheeseman J, et al. (2012) The effects of APOE on brain activity do not simply reflect the risk of Alzheimer’s disease. Neurobiol Aging. 33: 618.e1-618.e13. [crossref]
- Boisvert MM, Erikson GA, Shokhirev MN, Allen NJ (2018) The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep 22: 269-285. [crossref]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, et al. (2012) Genomic analysis of reactive astrogliosis. J Neurosci 32: 6391-6410. [crossref]
- Zhao N, Ren Y, Yamazaki Y, Qiao W, Li F, et al. (2020) Alzheimer’s risk factors age, APOE genotype, and sex drive distinct molecular pathways. Neuron 106: 727-742. [crossref]
- Dulewicz M, Kulczyska-Przybik A, Sowik A, Borawska R, Mroczko B (2021) Fatty acid binding protein 3 (FABP3) and apolipoprotein e4 (ApoE4) as lipid Metabolism-Related biomarkers of alzheimer’s disease. J Clin Med 10: 3009. [crossref]
- Rawat V, Wang S, Jian S, Bar R, Liraz O, et al. (2019) ApoE4 alters ABCA1 membrane trafficking in astrocytes. J Neurosci 39: 9611-9622. [crossref]
- Qiao F, Gao XP, Yuan L, Cai HY, Qi JS (2014) Apolipoprotein e4 impairs in vivo hippocampal Long-Term synaptic plasticity by reducing the phosphorylation of CaMKII伪 and CREB. Journal of Alzheimers Disease Jad 41: 1165-1176. [crossref]
- GA Rodriguez, MP, Burns, Weeber EJ, Rebeck GW (2013) Young APOE4 targeted replacement mice exhibit poor spatial learning and memory, with reduced dendritic spine density in the medial entorhinal cortex. Learn Memory 20: 256-266. [crossref]
- Terrisse L, Poirier J, Bertrand P, Merched A, Visvikis S, et al. (1998) Increased levels of apolipoprotein d in cerebrospinal fluid and hippocampus of alzheimer’s patients. J Neurochem 71: 1643-1650. [crossref]
- Lin YT, Seo J, Gao F, Feldman HM, Wen HL, et al. (2018) APOE4 causes widespread molecular and cellular alterations associated with alzheimer’s disease phenotypes in human iPSC-Derived brain cell types – ScienceDirect. Neuron 98: 1141-1154. [crossref]
- Sienski G, Narayan P, Bonner JM, Kory N, Boland S, et al. APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci Transl Med 13: eaaz4564. [crossref]
- Qi G, Mi Y, Shi X, Gu H, Brinton RD, et al. (2021) ApoE4 impairs Neuron-Astrocyte coupling of fatty acid metabolism. Cell Rep 34: 108572. [crossref]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, et al. (2014) An RNA-Sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34: 11929-11947. [crossref]
- Watkins PA (1997) Fatty acid activation. Progress in lipid research 36: 55-83.
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, et al. (2015) Tissue-based map of the human proteome. Science 347: 1260419. [crossref]
- Ellis JM, Frahm JL, Li LO, Coleman RA (2010) Acyl-coenzyme a synthetases in metabolic control. Curr Opin Lipidol 21: 212. [crossref]
- Steinberg SJ, Morgenthaler J, Heinzer AK, Smith KD, Watkins PA (2000) Very long-chain Acyl-CoA synthetases HUMAN “BUBBLEGUM” REPRESENTS a NEW FAMILY of PROTEINS CAPABLE of ACTIVATING VERY LONG-CHAIN FATTY ACIDS. J Biol Chem 275: 35162-35169. [crossref]
- Pei Z, Oey NA, Zuidervaart MM, Jia Z, Li Y, et al. (2003) The Acyl-CoA synthetase \”bubblegum\” (Lipidosin): FURTHER CHARACTERIZATION and ROLE in NEURONAL FATTY ACID? -OXIDATION. J Biol Chem 278: 47070-47078. [crossref]
- Li MD, Burns TC, Morgan AA, Khatri P (2014) Integrated multi-cohort transcriptional meta-analysis of neurodegenerative diseases. Acta Neuropathol Com 2: 1-23. [crossref]
- Jun G, Ibrahim-Verbaas CA, Vronskaya M, Lambert JC, Chung J, et al. (2016) A novel Alzheimer disease locus located near the gene encoding tau protein. Mol Psychiatry 21: 108-117. [crossref]
- Ellis JM, Bowman CE, Wolfgang MJ (2015) Metabolic and Tissue-Specific regulation of Acyl-CoA metabolism. Plos One 10: e0116587. [crossref]
- Marszalek JR, Kitidis C, Dararutana A, Lodish HF (2004) Acyl-CoA synthetase 2 overexpression enhances fatty acid internalization and neurite outgrowth. J Biol Chem 279: 23882-23891. [crossref]
- Klett EL, Chen S, Yechoor A, Lih FB, Coleman RA, et al. (2017) Long-chain acylCoA synthetase isoforms differ in preferences for eicosanoid species and long-chain fatty acids. J Lipid Res 58: 884-894. [crossref]
- Horn CGV, Caviglia JM, Li LO, Wang S, Granger DA, et al. (2005) Characterization of recombinant long-chain rat acyl-CoA synthetase isoforms 3 and 6: Identification of a novel variant of isoform 6. Biochemistry-Us 44: 1635-1642 [crossref]
- Kim HC, Lee SW, Cho YY, Lim JM, Ryoo ZY, et al. (2009) RNA interference of long-chain acyl-CoA synthetase 6 suppresses the neurite outgrowth of mouse neuroblastoma NB41A3 cells. Mol Med Rep 2: 669-674. [crossref]
- Wang X, Zhu M, Hjorth E, Cortés-Toro V, Eyjolfsdottir H, et al. (2015) Resolution of inflammation is altered in Alzheimer’s disease. Alzheimer’s & Dementia 11: 40-50. [crossref]
- Fernandez RF, Kim SQ, Zhao Y, Foguth RM, Weera MM, et al. (2018) Acyl-CoA synthetase 6 enriches the neuroprotective omega-3 fatty acid DHA in the brain. Proc Natl Acad Sci 115: 12525-12530. [crossref]
- Chouinard-Watkins R, Bazinet RP (2018) ACSL6 is critical for maintaining brain DHA levels. P Natl Acad Sci Usa 115: 12343-12345. [crossref]
- Fernandez RF, Pereyra AS, Diaz V, Wilson ES, Litwa KA, et al. (2021) Acyl-CoA synthetase 6 is required for brain docosahexaenoic acid retention and neuroprotection during aging. JCI Insight 6: e144351. [crossref]
- Fernandez RF, Ellis JM (2020) Acyl-CoA Synthetases as regulators of brain phospholipid acyl-chain diversity. Prostaglandins, Leukotrienes and Essential Fatty Acids. 161: 102175. [crossref]
- Sharma K, Schmitt S, Bergner CG, Tyanova S, Kannaiyan N, et al. (2015) Cell type- and brain region-resolved mouse brain proteome. Nat Neurosci 18: 1819-1831. [crossref]
- Atlas AMB (2009) Allen brain atlas. Neuron
- Orre M, Kamphuis W, Osborn LM, Melief J, Kooijman L, et al. (2014) Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiol Aging 35: 1-4. [crossref]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, et al. (2008) Transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J Neurosci 28: 264-278. [crossref]
- Soupene E, Kuypers FA (2008) Mammalian Long-Chain Acyl-CoA synthetases. Experimental Biology & Medicine 233: 507-521. [crossref]
- Hale BJ, Fernandez RF, Kim SQ, Diaz VD, Jackson SN, et al. (2019) Acyl-CoA Synthetase 6 enriches seminiferous tubules with the omega-3 fatty acid DHA and is required for male fertility in the mouse. J Biol Chem 294: 14394-14405. [crossref]
- Soupene E, Kuypers FA (2006) Multiple erythroid isoforms of human longchain acyl-CoA synthetases are produced by switch of the fatty acid gate domains. Bmc Mol Biol 7: 1-2. [crossref]
- Lee EJ, Kim HC, Yong YC, Byun SJ, Lim JM, et al. (2005) Alternative promotion of the mouse acyl-CoA synthetase 6 (mAcsl6) gene mediate the expression of multiple transcripts with 5′-end heterogeneity: Genetic organization of mAcsl6 variants. Biochem Biophys Res Commun 327: 84-93. [crossref]
- Malhotra KT, Malhotra K, Lubin BH, Kuypers FA (1999) Identification and molecular characterization of acyl-CoA synthetase in human erythrocytes and erythroid precursors. Biochem J 3441: 135-143. [crossref]
- Soupene E, Dinh NP, Siliakus M, Kuypers FA (2010) Activity of the acylCoA synthetase ACSL6 isoforms: Role of the fatty acid Gate-domains. Bmc Biochem 11: 1-3. [crossref]
- Gilquin B, Taillebourg E, Cherradi N, Hubstenberger A, Gay O, et al. (2010) The AAA+ ATPase ATAD3A controls mitochondrial dynamics at the interface of the inner and outer membranes. Molecular & Cellular Biology 30: 1984-96. [crossref]
- Desai R, Frazier AE, Durigon R, Patel H, Jones AW, et al. (2017) ATAD3 gene cluster deletions cause cerebellar dysfunction associated with altered mitochondrial DNA and cholesterol metabolism. Brain 140: 1595-1610. [crossref]
- Zhao Y, Sun X, Hu D, et al. (2019) ATAD3A oligomerization causes neurodegeneration by coupling mitochondrial fragmentation and bioenergetics defects. Nat Commun 10: 1-20.
- Harel T, Yoon WH, Garone C, Gu S, Coban-Akdemir Z, et al. (2016) Recurrent de novo and biallelic variation of ATAD3A, encoding a mitochondrial membrane protein, results in distinct neurological syndromes. Am J Hum Genet 99: 831-845. [crossref]
- Janikiewicz J, Szymański J, Malinska D, Patalas-Krawczyk P, Michalska B, et al. (2018) Mitochondria-associated membranes in aging and senescence: Structure,function, and dynamics. Cell Death Dis 9: 1-2. [crossref]
- Hayashi T, Rizzuto R, Hajnoczky G, Su TP (2009) MAM: More than just a housekeeper. Trends Cell Biol 19: 81-88. [crossref]
- Rusiol AE, Cui Z, Chen MH, Vance JE (1994) A unique mitochondria associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J Biol Chem 269: 27494-27502. [crossref]
- Vance JE (2003) Molecular and cell biology of phosphatidylserine and phosphatidylethanolamine metabolism. Prog Nucleic Acid Res Mol Biol 75: 69-111. [crossref]
- Mitsuo Tagaya, Kohei Arasaki (2017) Regulation of mitochondrial dynamics and autophagy by the Mitochondria-Associated membrane. Advances in Experimental Medicine & Biology 997: 33-47. [crossref]
- Csords G, Vrnai P, Golenr T, Roy S, Purkins G, et al. (2010) Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol Cell 39: 121-132. [crossref]
- Zhao Y, Hu D, Wang R, Sun X, Ropelewski P, et al. (2022) ATAD3A oligomerization promotes neuropathology and cognitive deficits in Alzheimer’s disease models. Nat Commun 13: 1-20. [crossref]
- Fathia Djelti, Braudeau J, Hudry E, Dhenain M, Varin J, et al. (2015) CYP46A1 inhibition, brain cholesterol accumulation and neurodegeneration pave the way for Alzheimer’s disease. Brain 138: 2383-2398. [crossref]
- Montesinos J, Pera M, Larrea D, Guardia-Laguarta C, Agrawal RR, et al. (2020) The Alzheimer’s disease associated C99 fragment of APP regulates cellular cholesterol trafficking. The EMBO Journal 39: e103791. [crossref]
- Weigel D, J Ckle H (1990) The fork head domain: A novel DNA binding motif of eukaryotic transcription factors? Cell. 63: 455-456. [crossref]
- Flachsbart F, Caliebe A, Kleindorp R, Blanché H, von Eller-Eberstein H, et al. (2009) Association of FOXO3Avariation with human longevity confirmed in German centenarians. Proceedings of the National Academy of Sciences 106: 2700-2705. [crossref]
- Willcox BJ, Portion TA, He Q, Chen R, Grove JS, et al. (2008) FOXO3A genotype is strongly associated with human longevity. Proceedings of the National Academy of Sciences 105: 13987-13992. [crossref]
- Shimokawa I, Komatsu T, Hayashi N, Kim SE, Kawata T, et al. (2015) The life-extending effect of dietary restriction requires Foxo3 in mice. Aging Cell 14: 707-709. [crossref]
- Chiacchiera F, Simone C (2010) The AMPK-FoxO3A axis as a target for cancer treatment. Cell cycle (Georgetown, Tex.) 9: 1091-1096. [crossref]
- Armando V, Burgering B (2007) Stressing the role of FoxO proteins in lifespan and disease. Nat Rev Mol Cell Biol 8: 440-450. [crossref]
- Beekman M, Nederstigt C, Suchiman H, Kramer D, Breggen R, et al. (2010) Genome-wide association study (GWAS)-identified disease risk alleles do not compromise human longevity. Proceedings of the National Academy of Sciences 107: 18046-18049. [crossref]
- Zettergren A, Kern S, Ryd茅n L, Blennow K, Zetterberg H, et al. (2018) Genetic variation in FOXO3 is associated with Self-Rated health in a Population-Based sample of older individuals. J Gerontol A Biol Med 73: 1453. [crossref]
- Soerensen M, Nygaard M, Dato S, Stevnsner T, Christiansen L (2015) Association study of FOXO3A SNPs and aging phenotypes in Danish oldest-old individuals. Aging Cell 14: 60-66. [crossref]
- Paik J, Ding Z, Narurkar R, Ramkissoon S, Muller F, et al. (2009) Fox Os cooperatively regulate diverse pathways governing neural stem cell homeostasis. Cell Stem Cell 5: 540-553. [crossref]
- Renault VM, Rafalski VA, Morgan AA, Salih DA, Brett JO, et al. (2009) FoxO3 regulates neural stem cell homeostasis. Cell Stem Cell 5: 527-539. [crossref]
- Schffner I, Minakaki G, Khan MA, Balta EA, Schlötzer-Schrehardt U, et al. (2018) FoxO function is essential for maintenance of autophagic flux and neuronal morphogenesis in adult neurogenesis. Neuron 99: 1188-1203. [crossref]
- Yeo H, Lyssiotis CA, Zhang Y, Ying H, Asara JM, et al. (2013) FoxO3 coordinates metabolic pathways to maintain redox balance in neural stem cells. Embo J 32: 2589-2602. [crossref]
- Caballero‐Caballero A, Engel T, Martinez‐Villarreal J, Sanz-Rodriguez A, Chang P, et al. (2013) Mitochondrial localization of the Forkhead box class O transcription factor FOXO3a in brain. J Neurochem 124: 749-756. [crossref]
- Lehtinen MK, Yuan Z, Boag PR, Yang Y, Villén J, et al. (2006) A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell 125: 987-1001. [crossref]
- Sriram K (2006) Deficiency of TNF receptors suppresses microglial activation and alters the susceptibility of brain regions to MPTP-induced neurotoxicity: Role of TNF-Faseb J 20: 670-682. [crossref]
- Woiciechowsky C, Sch枚ning B, Stoltenburg-Didinger G, Stockhammer F, Volk HD (2004) Brain-IL-1 beta triggers astrogliosis through induction of IL-6: Inhibition by propranolol and IL-10. Med Sci Monit 10: 325-330. [crossref]
- Buffo A, Rolando C, Ceruti S (2010) Astrocytes in the damaged brain: Molecular and cellular insights into their reactive response and healing potential. Biochem Pharmacol 79: 77-89. [crossref]
- Cui M, Huang Y, Tian C, et.al (2011) FOXO3a inhibits TNF– and IL-1-induced astrocyte proliferation: Implication for reactive astrogliosis. Glia 59: 641-654. [crossref]
- Du S, Jin F, Maneix L, et al. (2021) FoxO3 deficiency in cortical astrocytes leads to impaired lipid metabolism and aggravated amyloid pathology. Aging cell 20: e13432. [crossref]
- Cui M, Huang Y, Tian C, Zhao Y, Zheng J (2011) FOXO3a inhibits TNF– and IL-1-induced astrocyte proliferation: Implication for reactive astrogliosis. Glia 59: 641-654. [crossref]
- Nguyen TB, Louie SM, Daniele JR, Tran Q, Dillin A, et al. (2017) DGAT1-Dependent Lipid Droplet Biogenesis Protects Mitochondrial Function during Starvation-Induced Autophagy. Dev Cell 42: 9-21. [crossref]
- Ehehalt Robert, Keller P, Haass C, Thiele C, Simons K (2003) Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol 160: 113-123. [crossref]
- Saido T, Leissring MA (2012) Proteolytic degradation of amyloid beta-Protein. Csh Perspect Med 2: a6379. [crossref]
- He G, Luo W, Li P, Remmers C, Netzer WJ, et al. (2010) Gamma-secretase activating protein is a therapeutic target for Alzheimer’s disease. Nature 467: 95-98. [crossref]
- Chu J, Li JG, Joshi YB, Giannopoulos PF, Hoffman NE, et al. (2015) Gamma Secretase-Activating protein is a substrate for caspase-3: Implications for alzheimer’s disease. Biol Psychiat 77: 720-728. [crossref]
- Perez SE, Nadeem M, Malek-Ahmadi MH, He B, et al. (2017) Frontal cortex and hippocampal-Secretase activating protein levels in prodromal alzheimer disease. Neurodegener Dis 17: 235-241. [crossref]
- Satoh J, Tabunoki H, Ishida T, Saito Y, Arima K (2012) Immunohistochemical characterization of secretase activating protein expression in Alzheimer’s disease brains. Neuropathology & Applied Neurobiology 38: 132-141. [crossref]
- Floudas CS, Um N, Kamboh M, Barmada MM, Visweswaran S (2014) Identifying genetic interactions associated with late-onset Alzheimer’s disease. Biodata Min 7: 35. [crossref]
- Zhu M, Tao Y, He Q, Gao H, Song F, et al. (2014) A common GSAP promoter variant contributes to Alzheimer’s disease liability. Neurobiol Aging 35: 2651-2656. [crossref]
- Xu P, Chang JC, Zhou X, Wang W, Greengard P (2021) GSAP regulates lipid homeostasis and mitochondrial function associated with Alzheimer’s disease. The Journal of experimental medicine 218: e20202446. [crossref]
- Jin Chu, Elisabetta Lauretti, Caryne P, Praticò D (2014) Pharmacological modulation of GSAP reduces amyloid- levels and tau phosphorylation in a mouse model of Alzheimer’s disease with plaques and tangles. Journal of Alzheimers Disease Jad 41: 729-737. [crossref]
- Pera M, Larrea D, Guardia aguarta C, Montesinos J, Velasco KR, et al. (2017) Increased localization of APP C99 in mitochondria鈥恆ssociated ER membranes causes mitochondrial dysfunction in Alzheimer disease. The EMBO Journal. [crossref]
- Prete DD, Suski JM, Ouls B, Debayle D, Gay AS, et al. (2016) Localization and processing oftheAmyloid-beta protein precursor inMitochondria-Associated membranes. Journal of Alzheimer\”s Disease 55: 1549-1570. [crossref]
- Area-Gomez E, Maria DCLC, Tambini MD, Guardia-Laguarta C, De Groof AJC, et al. (2012) Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. Embo J 31: 4106-4123. [crossref]
- L Hedskog, CM Pinho, Filadi R, Rönnbäck A, Hertwig L, et al. (2013) Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proceedings of the National Academy of Sciences 110: 7916-7921. [crossref]
- Adami PVM, Nichtov Z, Weaver DB, Bartok A, Wisniewski T, et al. (2019) Perturbed mitochondriaR contacts in live neurons that model the amyloid pathology of Alzheimer’s disease. The Company of Biologists 132: jcs229906. [crossref]
- Kosicek M, Hecimovic S (2013) Phospholipids and alzheimer’s disease: Alterations, mechanisms and potential biomarkers. Int J Mol Sci 14: 1310-1322. [crossref]
- Goodenberger M, Jenkins RB (2012) Genetics of adult glioma. Cancer Genet-Ny 205: 613-621. [Crossref]
- Venur VA, Peereboom DM, Ahluwalia MS (2015) Current medical treatment of glioblastoma. Cancer Treatment & Research 163: 103-115. [crossref]
- Joo K, Kim J, Jin J, Kim M, Seol H, et al. (2013) Patient-Specific orthotopic glioblastoma xenograft models recapitulate the histopathology and biology of human glioblastomas in situ. Cell Rep [crossref]
- Ricard D, Idbaih A, Ducray F, Lahutte M, Hoangxuan K, et al. (2012) Primary brain tumours in adults. Lancet 379: 1984-1996. [crossref]
- Libby CJ, Tran AN, Scott SE, Griguer C, Hjelmeland AB (2018) The protumorigenic effects of metabolic alterations in glioblastoma including brain tumor initiating cells. Biochimica Et Biophysica Acta 1869: 175-188. [crossref]
- Agnihotri S, Zadeh G (2015) Metabolic reprogramming in glioblastoma: The influence of cancer metabolism on epigenetics and unanswered questions. Neuro Oncology 18: 160-172.[crossref]
- Intlekofer AM, Finley LWS (2019) Metabolic signatures of cancer cells and stem cells. Nature Metabolism 1: 177-188. [crossref]
- Weinberg H, Hanahan D (2011) Hallmarks of cancer: The next generation. Cell 144: 646-674. [crossref]
- Lopez-Lazaro M (2008) The warburg effect: Why and how do cancer cells activate glycolysis in the presence of oxygen? Anti-Cancer Agents in Medicinal. Chemistry (Formerly Current Medicinal Chemistry – Anti-Cancer Agents) 8. [crossref]
- Duraj T, Garcromero N, Carrinavarro J, Madurga R, Ana O, et al. Beyond the warburg effect: Oxidative and glycolytic phenotypes coexist within the metabolic heterogeneity of glioblastoma. Cells-Basel 10: 202. [crossref]
- Hoangnh LB, Siebzehnrubl FA, Yang C, Tano SS, Deleyrolle LP (2018) Infiltrative and drug鈥恟esistant slowcling cells support metabolic heterogeneity in glioblastoma. The EMBO Journal 37: e98772. [crossref]
- Caren Duman, Kaneschka Yaqubi, Angelika Hoffmann, Acikgöz AA, Korshunov A, et al. (2019) AcylCoA-Binding protein drives glioblastoma tumorigenesis by sustaining fatty acid oxidation. Cell Metab 30: 274-289. [crossref]
- Antonella DR, Serena P, Marco R, Patrizia T, Letizia M, et al. (2012) A radial glia gene marker, fatty acid binding protein 7 (FABP7), is involved in proliferation and invasion of glioblastoma cells. Plos One 7: e52113. [crossref]
- Gupta K, Vuckovic I, Zhang S, Xiong Y, Carlson BL, et al. (2020) Radiation induced metabolic alterations associate with tumor aggressiveness and poor outcome in glioblastoma. Frontiers Media S. A 10: 535. [crossref]
- Son B, Lee S, Kim H, Kang H, Youn BH (2020) Decreased FBP1 expression rewires metabolic processes affecting aggressiveness of glioblastoma. Oncogene 39: 36-49. [crossref]
- Hua L, Shaan P, Affleck VS, Ian W, Turnbull DM, et al. (2017) Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro-Oncology 43-54. [crossref]
- Hanna VS, Ebtisam A (2018) Synopsis of arachidonic acid metabolism: A review. J Adv Res 23-32. [crossref]
- Juric-Sekhar G, Zarkovic K, Waeg G, Cipak A, Zarkovic N (2009) Distribution of 4-hydroxynonenal-protein conjugates as a marker of lipid peroxidation and parameter of malignancy in astrocytic and ependymal tumors of the brain. Tumori 95: 762-768. [crossref]
- Zajdel A, Wilczok A, Slowinski J, Mazurek OU (2007) Aldehydic lipid peroxidation products in human brain astrocytomas. J Neuro-Oncol 84: 167-174. [crossref]
- Zarkovic K, Juric G, Waeg G, Kolenc D, Zarkovic N (2010) Immunohistochemical appearance of HNE-protein conjugates in human astrocytomas. Biofactors 24: 33-40. [crossref]
- Atilla-Gokcumen G, Muro E, Relat-Goberna J, Sasse S, Bedigian A, Coughlin M, et al. (2014) Dividing cells regulate their lipid composition and localization. Cell 156. [crossref]
- Lopez DH, Bestard-Escalas J, Garate J, Maim贸-Barcel贸 A, Fern谩ndez R, Reigada R, et al. (2018) Tissue-selective alteration of ethanolamine plasmalogen metabolism in dedifferentiated colon mucosa. Biochimica et Biophysica Acta (BBA) – Molecular and Cell Biology of Lipids:S263724189. [crossref]
- Sang TL, Lee JC, Kim JW, Cho SY, Seong JK, Moon MH (2016) Global changes in lipid profiles of mouse cortex, hippocampus, and hypothalamus upon p53 knockout. Sci Rep-Uk 6: 36510. [crossref]
- Baenke F, Peck B, Miess H, Schulze A (2013) Hooked on fat: The role of lipid synthesis in cancer metabolism and tumour development. Dis Model Mech 6: 1353-1363. [crossref]
- Gimple RC, Kidwell RL, Ljy K, Sun T, Gromovsky AD, et al. (2019) Glioma stem cell specific super enhancer promotes polyunsaturated fatty acid synthesis to support EGFR signaling. Cancer Discov. [crossref]
- Zou YK, Watters A, Cheng N, Perry CE, Chen Q (2019) Polyunsaturated fatty acids from astrocytes activate PPARγ signaling in cancer cells to promote brain metastasis. Cancer Discov 9: 1720-1735. [crossref]
- Cheng X, Geng F, Pan M, Wu X, Guo D (2020) Targeting DGAT1 ameliorates glioblastoma by increasing fat catabolism and oxidative stress. Cell Metab [crossref]
- Omuro A, Deangelis LM (2013) Glioblastoma and other malignant gliomas: A clinical review. Journal of the American Medical Association 310: 1842-1850. [crossref]
- Wen PY, Kesari S (2008) Malignant gliomas in adults. New Engl J Med 359: 492-507. [crossref]
- Accioly MT, Pacheco P, Maya-Monteiro CM, Carrossini N, Robbs BK, et al. (2008) Lipid bodies are reservoirs of cyclooxygenase-2 and sites of Prostaglandin-E2 synthesis in colon cancer cells. Cancer Res 68: 1732-1740. [crossref]
- Du W, Zhang L, Brett-Morris A, Aguila B, Kerner J, et al. (2017) HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat Commun 8: 1769. [crossref]
- Feng G, Xiang C, Wu X, Ji YY, Guo D (2017) Inhibition of SOAT1 suppresses glioblastoma growth via blocking SREBP-1-mediated lipogenesis. Clin Cancer Res 22. [crossref]
- Geng F, Guo D (2017) Lipid droplets, potential biomarker and metabolic target in glioblastoma. Intern Med Rev 3: 10. [crossref]
- Mitra R, Le TT, Gorjala P, Goodman OB (2017) Positive regulation of prostate cancer cell growth by lipid droplet forming and processing enzymes DGAT1 and ABHD5. Bmc Cancer 17: 631. [crossref]
- Pucer A, Brglez V, Payr C, Pungerar J, Petan T (2013) Group X secreted phospholipase A2 induces lipid droplet formation and prolongs breast cancer cell survival. Mol Cancer 12: 111. [crossref]
- Sevinsky CJ, Faiza K, Leila K, Anza D, Rao MK, et al. (2018) NDRG1 regulates neutral lipid metabolism in breast cancer cells. Breast Cancer Res 20: 55. [crossref]
- Sunami Y, Rebelo A, Kleeff J (2017) Lipid metabolism and lipid droplets in pancreatic cancer and stellate cells. Cancers 10: 3. [crossref]
- Tauchi-Sato K, Ozeki S, Houjou T, Taguchi R, Fujimoto T (2002) The surface of lipid droplets is a phospholipid monolayer with a unique fatty acid composition. J Biol Chem 277: 44507-44512. [crossref]
- Wilfling F, Haas JT, Walther TC, Jr R (2014) Lipid droplet biogenesis. Curr Opin Cell Biol [crossref]
- Olzmann James A, Carvalho Pedro (2019) Dynamics and functions of lipid droplets. 20: 137-155. [crossref]
- Paar M, Jungst C, Steiner NA, Magnes C, Sinner F, et al. (2012) Remodeling of lipid droplets during lipolysis and growth in adipocytes. J Biol Chem 287: 11164-11173. [crossref]
- Klionsky DJ, Emr SD (2000) Autophagy as a regulated pathway of cellular degradation. Science 290: 1717-1721. [crossref]
- Mizushima N, Komatsu M (2011) Autophagy: Renovation of cells and tissues. Cell 147: 728-741. [crossref]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self-digestion. Nature 451: 1069. [crossref]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, et al. (2009) Autophagy regulates lipid metabolism. Nature 458: 1131-1135. [crossref]
- Wu X, Geng F, Cheng X, Guo Q, Zhong Y, et al. (2020) Lipid droplets maintain energy homeostasis and glioblastoma growth via autophagic release of stored fatty acids. iScience 23: 101569. [crossref]
- Guo D, Reinitz F, Youssef M, Hong C, Nathanson D, Akhavan D, et al. (2011) An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov 1: 442-456. [crossref]
- Bjorkhem I (2004) Brain cholesterol: Long secret life behind a barrier. Arteriosclerosis, Thrombosis, and Vascular Biology 24: 806-815. [crossref]
- Dietschy JM, Turley SD (2001) Cholesterol metabolism in the brain. Curr Opin Lipidol 12: 105-112. [crossref]
- Hayashi H, Campenot RB, Vance DE, Vance JE (2004) Glial lipoproteins stimulate axon growth of central nervous system neurons in compartmented cultures. J Biol Chem 279: 14009-14015. [crossref]
- Karten B, Campenot RB, Vance DE, Vance JE (2006) Expression of ABCG1, but not ABCA1, correlates with cholesterol release by cerebellar astroglia. J Biol Chem 281: 4049-4057. [crossref]
- Wahrle SE, Jiang H, Parsadanian M, Legleiter J, Han X, et al. (2004) ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J Biol Chem 279: 40987-40993. [crossref]
- Orth M, Bellosta S (2012) Cholesterol: Its regulation and role in central nervous system disorders. Cholesterol 2012: 292598. [crossref]
- Jing C, Zhang X, Kusumo H, Costa LG, Guizzetti M (2013) Cholesterol efflux is differentially regulated in neurons and astrocytes: Implications for brain cholesterol homeostasis. Biochimica et Biophysica Acta (BBA) – Molecular and Cell Biology of Lipids 1831: 263-275. [crossref]
- Repa JJ (2000) Regulation of absorption and ABC1-Mediated efflux of cholesterol by RXR heterodimers. Science 289: 1524-1529. [crossref]
- Venkateswaran Asha, Laffitte Bryan A (2000) Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha. P Natl Acad Sci Usa 97: 12097-12102. [crossref]
- Zelcer N, Hong C, Boyadjian R, Tontonoz P (2009) LXR regulates cholesterol uptake through Idol-Dependent ubiquitination of the LDL receptor. Science 325: 100-104. [crossref]
- Villa G, Hulce J, Zanca C, Bi J, Mischel P (2016) An LXR-Cholesterol axis creates a metabolic Co-Dependency for brain cancers. Cancer Cell 30: 683-693. [crossref]
- Friedman HS, Kerby T, Calvert H (2000) Temozolomide and treatment of malignant glioma. Clinical cancer research: an official journal of the American Association for Cancer Research 6: 2585-2597. [crossref]
- Stupp R, Mason WP, Van D, Weller M, Fisher B, et al. (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma – Cancer/Radiothrapie 9: 196-197. [crossref]
- Massimo Aureli, Valentina Murdica, Nicoletta Loberto, Samarani M, Prinetti A, et al. (2014) Exploring the link between ceramide and ionizing radiation. Glycoconjugate J 31: 449-459. [crossref]
- Brady RO, Kanfer JN, Mock MB, Fredrickson DS (1966) The metabolism of sphingomyelin. II. Evidence of an enzymatic deficiency in Niemann-Pick diseae. Proceedings of the National Academy of Sciences 55: 366-369. [crossref]
- Haimovitz-Friedman A (1994) Ionizing radiation acts on cellular membranes to generate ceramide and initiate apoptosis. J Exp Med 180: 525-535. [crossref]
- Senchenkov A, Litvak DA, Cabot MC (2001) Targeting ceramide metabolism strategy for overcoming drug resistance. JNCI J Natl Cancer Inst 93: 347-357. [crossref]
- Mizushima N, Koike R, Kohsaka H, Kushi Y, Handa S, et al. (1996) Ceramide induces apoptosis via CPP32 activation. Febs Lett 395: 261-271. [crossref]
- Abuhusain HJ, Matin A, Qiao Q, Shen H, Kain N, et al. (2013) A metabolic shift favoring sphingosine 1-Phosphate at the expense of ceramide controls glioblastoma angiogenesis. J Biol Chem 288: 37355-37364. [crossref]
- Ji-Sun J, Young-Ho A, Byung-In M, Hee-Sun K (2016) Exogenous c2 ceramide suppresses matrix metalloproteinase gene expression by inhibiting ROS production and MAPK signaling pathways in PMA-Stimulated human astroglioma cells. Int J Mol Sci 17: 477. [crossref]
- Nganga R, Oleinik N, Ogretmen B (2018) Mechanisms of Ceramide-Dependent cancer cell death. Adv Cancer Res [crossref]
- Gault CR, Obeid LM, Hannun YA (2010) An overview of sphingolipid metabolism: From synthesis to breakdown. Oxygen Transport to Tissue XXXIII 688: 1-23. [Crossref]
- Buehrer BM, Bell RM (1992) Inhibition of sphingosine kinase in vitro and in platelets. Implications for signal transduction pathways. J Biol Chem 267: 3154-3159. [crossref]
- Brocklyn J, Young N, Roof R (2003) Sphingosine-1-phosphate stimulates motility and invasiveness of human glioblastoma multiforme cells. Cancer Lett 199: 53-60. [crossref]
- Bassi R, Anelli V, Giussani P, Tettamanti G, Viani P, et al. (2010) Sphingosine-1-phosphate is released by cerebellar astrocytes in response to bFGF and induces astrocyte proliferation through Gi-protein-coupled receptors. Glia 53: 621-630. [crossref]
- Anelli V, Gault CR, Cheng AB, Obeid LM (2008) Sphingosine kinase 1 is up-regulated during hypoxia in U87MG glioma cells. J Biol Chem 283: 3365-3375. [crossref]
- Strub GM, Maceyka M, Hait NC, Milstien S, Spiegel S (2010) Extracellular and intracellular actions of Sphingosine-1-Phosphate. Adv Exp Med Biol 688: 141-155. [crossref]
- Cuvillier O, Pirianov G, Kleuser B, Vanek PG, Spiegel S (1996) Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 381: 800-803. [crossref]
- Masayuki Nagahashi, Manabu Abe, Kenji Sakimura, Takabe K, Wakai T (2018) The role of sphingosine-1-phosphate in inflammation and cancer progression. Cancer Sci 109: 3671-3678. [crossref]
- Marfia G, Campanella R, Navone SE, Vito CD, Riccitelli E, et al. (2014) Autocrine/paracrine sphingosine phosphate fuels proliferative and stemness qualities of glioblastoma stem cells. Glia 62: 1968-1981. [crossref]
- Im DS, Clemens J, Macdonald TL, Lynch KR (2001) Characterization of the human and mouse sphingosine 1-phosphate receptor, S1P5 (Edg-8): Structure-activity relationship of sphingosine1-phosphate receptors. Biochemistry-Us 40: 14053-14060. [crossref]
- Hla T, Maciag T (1990) An abundant transcript induced in differentiating human endothelial cells encodes a polypeptide with structural similarities to Gprotein-coupled receptors. J Biol Chem 265: 9308-9313. [crossref]
- Masana MI, Brown RC, Pu H, Gurney ME, Dubocovich ML (1995) Cloning and characterization of a new member of the G-protein coupled receptor EDG family. Receptors Channels 3: 255-262. [crossref]
- Zondag GC, Postma FR, Etten IV, Verlaan I, Moolenaar WH (1998) Sphingosine 1-phosphate signalling through the G-protein-coupled receptor Edg-1. The Biochemical journal 330: 605-609. [crossref]
- Siehler S, Manning DR (2002) Pathways of transduction engaged by sphingosine 1-phosphate through G protein-coupled receptors. Biochimica et Biophysica Acta 1582: 94-99. [crossref]
- Sanchez T, Hla T (2004) Structural and functional characteristics of S1P receptors. J Cell Biochem 92. [crossref]
- Bien-Miller S, Lange S, Holm T, B枚hm A, Rauch BH (2016) Expression of S1P metabolizing enzymes and receptors correlate with survival time and regulate cell migration in glioblastoma multiforme. Oncotarget 7. [crossref]
- Yoshida Y, Nakada M, Sugimoto N, Harada T, Hayashi Y, et al. (2010) Sphingosine-1-phosphate receptor type 1 regulates glioma cell proliferation and correlates with patient survival. Int J Cancer 126: 2341-2352. [crossref]
- Bernhart E, Damm S, Wintersperger A, Nusshold C, Brunner AM, et al. (2015) Interference with distinct steps of sphingolipid synthesis and signaling attenuates proliferation of U87MG glioma cells. Biochem Pharmacol [crossref]
- Young N, Brocklyn JRV (2007) Roles of Sphingosine-1-Phosphate (S1P) receptors in malignant behavior of glioma cells. Differential effects of S1P2 on cell migration and invasiveness. Exp Cell Res 313: 1615-1627. [crossref]
- Lepley D (2005) The g protein鈥揅oupled receptor S1P2 regulates Rho/Rho kinase pathway to inhibit tumor cell migration. Cancer Res 65: 3788. [crossref]
- Malchinkhuu E, Sato K, Maehama T, Mogi C, Tomura H, Ishiuchi S, et al. (2008) S1P(2) receptors mediate inhibition of glioma cell migration through Rho signaling pathways independent of PTEN. Biochem Biophys Res Commun 366(4):963-968 [crossref]
- Quint K, Stiel N, Neureiter D, Schlicker HU, Nimsky C, et al. (2014) The role of sphingosine kinase isoforms and receptors S1P1, S1P2, S1P3, and S1P5 in primary, secondary, and recurrent glioblastomas. Tumour Biol 35: 8979-8989. [crossref]
- Rothhammer V, Kenison JE, Tjon E, Takenaka MC, Lima KD, et al. (2017) Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proceedings of the National Academy of ences 114: 201615413. [crossref]
- Sullivan SA, O Sullivan C, Healy LM, Dev KK, Sheridan GK (2018) Sphingosine 1鈥恜hosphate receptors regulate TLR4 innduced CXCL5 release from astrocytes and microglia. J Neurochem 144: 736-747. [crossref]
- Dai Z, Wu J, Chen F, Cheng Q, Zhang M, et al. (2019) Cxcl5 promotes the proliferation and migration of glioma cells in autocrine-and paracrinedependent manners. Oncol Rep 36: 3303-3310. [crossref]
- Windh RT, Lee MJ, Hla T, An S, Barr AJ, et al. (1999) Differential coupling of the sphingosine 1-Phosphate receptors edg-1, edg-3, and H218/Edg-5 to the gi, gq, and g12 families of heterotrimeric g proteins. J Biol Chem 274: 27351-27358. [crossref]
- Liu Y, Wang X, Li J, Tang J, Li B, et al. (2021) Sphingosine 1-Phosphate liposomes for targeted nitric oxide delivery to mediate anticancer effects against brain glioma tumors. Adv Mater.
- Bien-M枚ller S, Lange S, Holm T, B枚hm A, Rauch BH (2016) Expression of S1P metabolizing enzymes and receptors correlate with survival time and regulate cell migration in glioblastoma multiforme. Oncotarget 7. [crossref]
- Rekers NH, Sminia P, Peters GJ (2011) Towards tailored therapy of glioblastoma multiforme. J Chemotherapy 23: 187-199. [crossref]
- Bryan L, Paugh BS, Kapitonov D, Wilczynska KM, Alvarez SM, et al. (2008) Sphingosine-1-phosphate and interleukin-1 independently regulate plasminogen activator inhibitor-1 and urokinase-type plasminogen activator receptor expression in glioblastoma cells: Implications for invasiveness. Molecular Cancer Research Mcr 6: 1469. [crossref]
- Young N, Brocklyn JRV (2007) Roles of Sphingosine-1-Phosphate (S1P) receptors in malignant behavior of glioma cells. Differential effects of S1P2 on cell migration and invasiveness. Exp Cell Res 313: 1615-1627. [crossref]
- Sato K, Tomura H, Igarashi Y, Ui M, Okajima F (1999) Possible involvement of cell surface receptors in sphingosine 1-phosphate-induced activation of extracellular signal-regulated kinase in C6 glioma cells. Mol Pharmacol 55: 126-133. [crossref]
- Malchinkhuu E, Sato K, Horiuchi Y, Mogi C, Ohwada S, et al. (2005) Role of p38 mitogen-activated kinase and c-Jun terminal kinase in migration response to lysophosphatidic acid and sphingosine-1-phosphate in glioma cells. Oncogene 24: 6676-6688. [crossref]
- Brocklyn JRV, Young N, Roof R (2003) Sphingosine-1-phosphate stimulates motility and invasiveness of human glioblastoma multiforme cells. Cancer Lett 199: 53-60. [crossref]
- Yoshida Y, Nakada M, Harada T, Tanaka S, Furuta T, et al. (2010) The expression level of sphingosine-1-phosphate receptor type 1 is related to MIB-1 labeling index and predicts survival of glioblastoma patients. J Neuro-Oncol 98: 41-47. [crossref]
- Yoshida Y, Nakada M, Sugimoto N, Harada T, Hayashi Y, et al. (2010) Sphingosine-1-phosphate receptor type 1 regulates glioma cell proliferation and correlates with patient survival. Int J Cancer 126: 2341-2352. [crossref]
- Shimizu F, Watanabe TK, Shinomiya H, Nakamura Y, Fujiwara T (1997) Isolation and expression of a cDNA for human brain fatty acid-binding protein (BFABP). Biochimica et Biophysica Acta (BBA) – Gene Structure and Expression 1354: 24-28. [crossref]
- Xu LZ, Sanchez R, Sali A, Heintz N (1996) Ligand specificity of brain lipidbinding protein. J Biol Chem 271: 24711. [crossref]
- Kurtz A, Zimmer A, Schngen F, Brning G, Miller T (1994) The expression pattern of a novel gene encoding brain-fatty acid binding protein correlates with neuronal and glial cell development. Development 120: 2637-2649. [crossref]
- Owada Y, Yoshimoto T, Kondo H (1996) Spatio-temporally differential expression of genes for three members of fatty acid binding proteins in developing and mature rat brains. J Chem Neuroanat 12: 113-122. [crossref]
- Yun SW, Leong C, Zhai D, Tan YL, Lim L, et al. (2012) Neural stem cell specific fluorescent chemical probe binding to FABP7. P Natl Acad Sci Usa 109: 10214-10217.
- Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, et al. (2003) Identification of a cancer stem cell in human brain tumors. Cancer Res 63: 5821. [crossref]
- Llaguno SA, Chen J, Parada LF (2009) Signalling in malignant astrocytomas: Role of neural stem cells and its therapeutic implications. Clinical Cancer Research An Official Journal of the American Association for Cancer Research 15: 7124-7129. [crossref]
- Kaloshi G, Mokhtari K, Carpentier C, Sophie T, Lejeune J, et al. (2007) FABP7 expression in glioblastomas: Relation to prognosis, invasion and EGFR status. J Neuro-Oncol 84: 245-248. [crossref]
- Liang YU, Maximilian D, Nathan W, Bollen AW, Aldape KD, et al. (2005) Gene expression profiling reveals molecularly and clinically distinct subtypes of glioblastoma multiforme. Proc Natl Acad Sci USA 102: 5814-5819. [crossref]
- Liang Y, Bollen AW, Aldape KD, Gupta N (2006) Nuclear FABP7 immunoreactivity is preferentially expressed in infiltrative glioma and is associated with poor prognosis in EGFR-overexpressing glioblastoma. Bmc Cancer 6: 97. [crossref]
- Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, et al. (2014) Fatty acid uptake and lipid storage induced by HIF-1伪 contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep 9: 349-365.
- Hoangnh LB, Siebzehnrubl FA, Yang C, Tano SS, Deleyrolle LP (2018) Infiltrative and drug resistant slowcling cells support metabolic heterogeneity in glioblastoma. The EMBO Journal 37: e98772. [crossref]
- Xu X, Wang Y, Choi WS, Sun X, Godbout R (2021) Super resolution microscopy reveals DHA-dependent alterations in glioblastoma membrane remodelling and cell migration. Nanoscale 21.
- Paton CM, Ntambi JM (2008) Biochemical and physiological function of stearoyl-CoA desaturase. The American journal of physiology 297: E28-E37. [crossref]
- Matthew T Flowers, James M Ntambi (2008) Role of stearoyl-coenzyme a desaturase in regulating lipid metabolism. Curr Opin Lipidol 19: 248-256. [crossref]
- Wu X, Zou X, Chang Q, Zhang Y, Li Y, et al. (2013) The evolutionary pattern and the regulation of Stearoyl-CoA desaturase genes. Biomed Res Int 2013: 856521. [crossref]
- Ecker J, Liebisch G, Grandl M, Schmitz G (2010) Lower SCD expression in dendritic cells compared to macrophages leads to membrane lipids with less monounsaturated fatty acids. Immunobiology 215: 748-755. [crossref]
- Zhang L, Ge L, Parimoo S, Stenn K, Prouty SM (1999) Human stearoyl-CoA desaturase: Alternative transcripts generated from a single gene by usage of tandem polyadenylation sites. Biochem J 340: 255-264. [crossref]
- Noushmehr H, Weisenberger D, Diefes K, Phillips H, Pujara K, et al. (2010) The cancer genome atlas research network. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17: 510-522. [crossref]
- Menendez J, Lupu R (2007) Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 7: 763-777. [Crossref]
- Peck, Barrie, Schulze, Almut. (2016) Lipid desaturation – the next step in targeting lipogenesis in cancer? Febs J 283: 2767-2778. [crossref]
- Hilvo M, Denkert C, Lehtinen L, Muller B, Brockmoller S, et al. (2011) Novel theranostic opportunities offered by characterization of altered membrane lipid metabolism in breast cancer progression. Cancer Res 71: 3236-3245. [crossref]
- Scaglia N, Caviglia JM, Igal RA (2005) High stearoyl-CoA desaturase protein and activity levels in simian virus 40 transformed-human lung fibroblasts. Biochim Biophys Acta 1687: 141-151. [crossref]
- Brennan CW, Verhaak R, Mckenna A, Campos B, Noushmehr H, et al. (2013) The somatic genomic landscape of glioblastoma. Cell 155: 462-477. [crossref]
- Oatman N, Dasgupta N, Arora P, Choi K, Dasgupta B (2021) Mechanisms of stearoyl CoA desaturase inhibitor sensitivity and acquired resistance in cancer. Sci Adv 7: d7459. [crossref]