DOI: 10.31038/CST.2020513
Heparan sulfate (HS) is a sulfated glycosaminoglycan that is deposited in human tissue matrices at specialized sites [1,2]. HS interacts with diverse extracellular matrix (ECM) components with HS binding sites, including inflammatory cytokines, and heparin-binding growth factors (HBGFs) [3,4]. Within the ECM and in the cell surface glycocalyx, HS-proteoglycans (HSPGs) act as reservoirs for cytokines and HBGFs, and as cofactors for surface receptors where they stabilize active signaling complexes [5–7]. The bioavailability and activity of HBGFs stored on HSPGs are primarily regulated by HS-modifying enzymes that act on HSPGs, such as perlecan, the syndecans and the glypicans [8,9]. Therefore, HSPGs and their enzymic modifiers are crucial for tissue homeostasis, both in normal biology, as in development and wound healing, and in pathological processes such as fibrosis and cancer biology [1,10,11]. To date, studies have identified three key extracellular enzymes that modulate HS function and growth factor signaling: tissue heparanase (HPSE) and the extracellular endosulfatases SULF1 and SULF2. HPSE is an endoglycosidase that cleaves HS chains yielding diffusible HS fragments [12] that often still retain bound growth factors (Fig. 1A). HS-bound growth factors can subsequently bind to surface receptors to form HS-HBGF-receptor ternary complexes (Fig. 1B) [12]. Like HPSE, SULFs are secreted but, for the most part, stay peripherally associated with the cell surface through the interaction with HSPGs in the glycocalyx, primarily syndecans and glypicans [13,14]. Enzymatic activity of SULFs involves selectively removing 6-O-sulfate groups from HS polymers (Figure 1A) [14,15]. Because many HBGFs require 6-O-sulfate for high-affinity binding to HSPGs or surface coreceptors [3,15,16], SULFs release HBGFs in a form free from HS chains. Freed HBGFs can bind subsequently to cognate cell surface receptors to form signaling complexes, or they may rebind to distant unmodified HSPGs that retain 6-O-sulfate. Therefore, both HPSE and SULFs are crucial enzymes that define activation parameters of HS-independent signaling networks in both positive and negative ways that often are context-dependent [17,18].
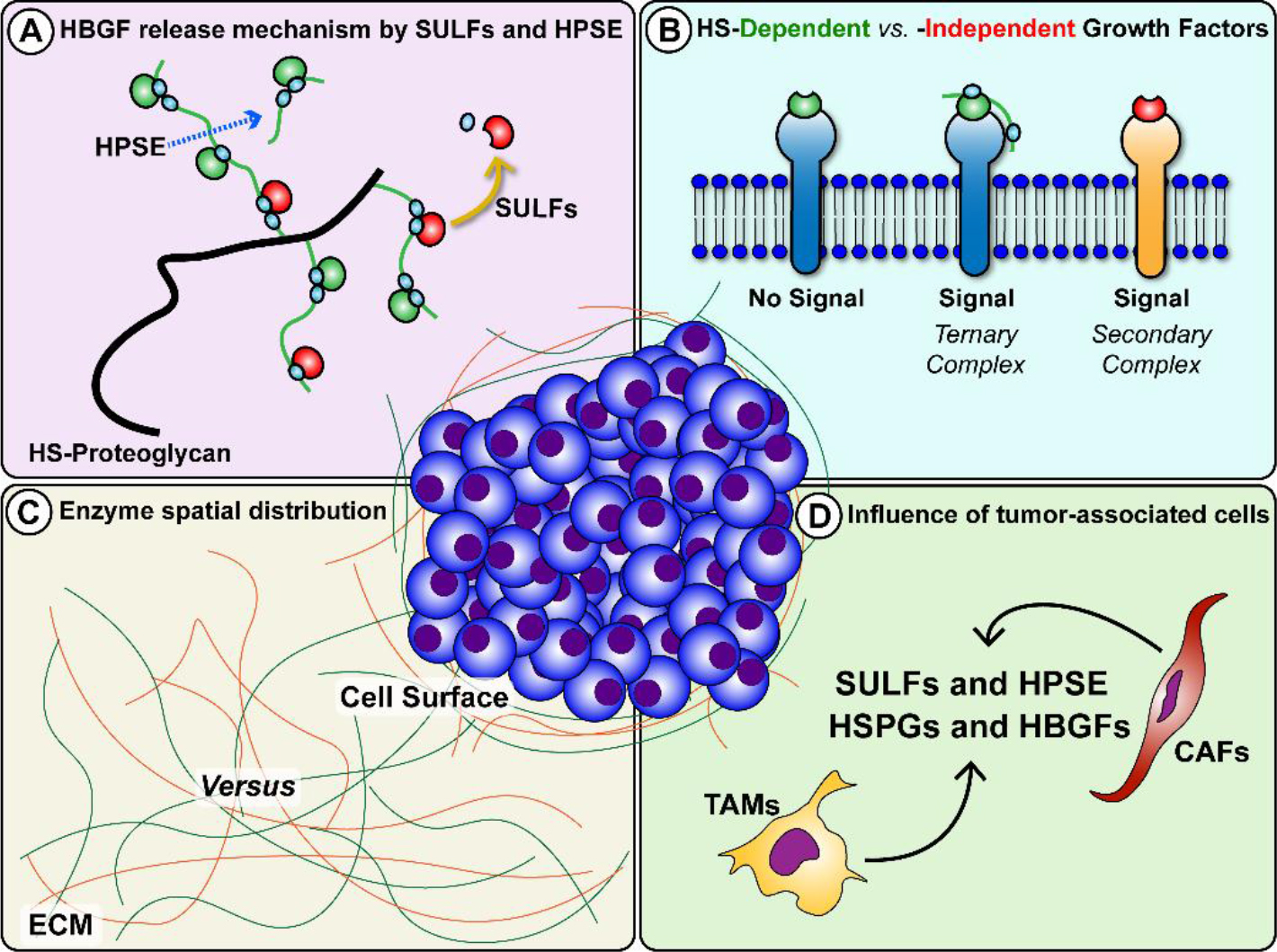
Figure 1. HS-modifying enzymes HPSE and SULFs release HBGFs with outcomes that are influenced by context. A. HPSE directly cuts HS chains to increase availability of HBGFs bound to HS fragments, while SULFs remove 6-O-sulfate residues (light blue circles) and release HBGFs free of HS. B. HS can act as a cofactor and stabilize HBGF binding to receptors via ternary complexes, while other factors transduce their signals via binary complexes. C. Spatial distribution of HPSE and SULFs produce opposing signaling effects when these enzymes act at the cell surface versus release factors bound in the ECM. At the cell surface, SULFs can disrupt ternary complex formation by HS desulfation, inhibiting downstream signaling. D. Infiltration and activation of tumor-associated cells, both TAMs and CAFs, contribute to regulation of HPSE and SULF expression and enrich the tumor milieu with HSPGs and HBGFs.
Better understood than the SULFs, HPSE generally is regarded as a tumor promoter. Cleavage of HS by HPSE releases and increases the availability of HBGFs, including vascular endothelial growth factors, hepatocyte growth factors [19–22] and fibroblast growth factors [23–26], thereby improving their access to their cell surface receptors and enabling downstream growth signaling. Consequently, HPSE can stimulate pro-tumorigenic processes including neoangiogenesis, tumor cell proliferation and invasion, inhibition of apoptosis, and metastasis, all among the well-accepted hallmarks of cancer [27,28]. Because of the intricacies from potential outcomes of SULF activity, predicting their impact on complex microenvironments, a priori, such as tumors, is more complicated. Numerous studies have implicated the SULFs as significant players involved in critical aspects of cancer progression, including proliferation, invasion and metastasis [1,15]. The expression of these intriguing enzymes is abnormal in many carcinoma cells, yet no consensus conclusion has been made as to whether they support or inhibit general cancer progression. Some of this confusion may be attributed to differences in regulation of gene expression between SULF1 and SULF2. For example, tumor necrosis factor α (TNFα) [29] and Wilm’s tumor transcriptional factor [30] stimulate SULF1 expression to a greater extent than SULF2. In contrast, SULF2, but not SULF1, is a p53 target [31]. A comparison of potential transcription factor binding sites (TFBS) in the SULF1 and SULF2 promoter regions in silico revealed that ~50% of TBFS were not shared between these two genes [32]. Therefore, dysregulated transcriptional programs and different transcriptional targeting in SULF genes both in cancer cells and cells in the tumor microenvironment may partially explain some of the apparently contradicting data concerning SULF functions in tumorigenesis.
A review of studies focusing on SULFs and published in the past twenty years reveals contrasting expression levels and opposing effects on tumor growth depending on the type of cancer and the surrounding microenvironment. For instance, an analysis of SULF1/SULF2 in various cancer cell lines suggested a mostly tumor-suppressing role of SULFs [33]. In contrast, other researchers demonstrated that high SULF1 or SULF2 levels correlate with poor prognosis in a wide range of tumor types [34]. Additionally, contrary to SULF2, SULF1 can exert a tumor suppressor effect in cancers, including myeloma, ovarian, head and neck, breast, liver, and pancreatic [33,35–39] cancers, despite being upregulated in others [40]. The paradox of how SULFs, sharing essentially identical target specificity, have different biological functions remains an open research question. In seeking to reconcile these observations, an essential point to consider is the signaling context. Most of the studies mentioned above solely focused on the cancer compartment, where cultured cells respond to artificially supplied HBGFs. However, there is overwhelming evidence that associated “bystander” stromal cells play a vital role in the regulation of tumor growth [41–13]. Cancers with reduced expression of HPSE or the SULFs still may be impacted by the actions of these enzymes in scenarios where they are being produced by cancer-associated fibroblasts (CAFs) and/or tumor-associated macrophages (TAMs). In recent years, the role of immune cells in cancer progression has gained increased attention. TAMs stand out as a major cell population in the tumor stroma [44] where they can, together with CAFs, modulate the expression of matrix remodeling enzymes, HSPGs, and HBGFs via pro- and anti-inflammatory cytokines [45–48] (Fig. 1D).
Also part of the signaling context controlling cell behavior are the specific ligands and their binding preferences to various HS modifications, spatial distribution of the enzymes themselves, cellular composition of the microenvironment, and the combination of HBGFs and cytokines present. Examples of such variations include whether: 1) ligands require HS fragments as cofactors for ternary complex signaling (Fig. 1B); 2) desulfation results in HBGF release or disruption of cofactor potential; 3) the enzymes are more abundant at the cell surface or in the ECM (Fig. 1C); 4) a robust reactive stroma response supporting cancer progression is present. While SULFs have been shown to suppress signaling at the cell surface through disruption of coreceptor functions, their release of HBGFs from fibroblasts in a desmoplastic stroma might favor growth. To date, studies exploring the influence of these different aspects of the signaling context are scarce, primarily from a lack of in vitro model systems that can reproduce the convoluted tumor microenvironment. Recent improvements in bioengineered cancer tissues are changing this, and new insights are on the horizon. While several HPSE inhibitors have reached and/or are currently undergoing clinical trials [49,50], no drug targeting the SULFs specifically has reached the clinic. Given the diverse nature of SULF expression and opposing activity in different contexts, as discussed above, targeting SULFs for cancer therapy is a complex endeavor. A key concern relates to the consequences of potentiating or inhibiting SULF activity. While silencing SULFs can lead to anti-tumor effects in some cancers, in others where they act as tumor suppressors, SULF inhibition could enhance tumorigenicity. A significant amount of pre-clinical work is needed to understand the full repertoire of pro- and anti-tumor activities of the SULFs such that SULF-based therapies can be designed with confidence. Nonetheless, the undeniable involvement of HPSE and SULFs in regulating cancer progression makes these enzymes attractive both as therapeutic targets and prognostic indicators of tumor progression.
Acknowledgements
This work was supported by P01CA098912 from the National Institutes of Health and the Brazilian Coordination for the Improvement of Higher Education Personnel (CAPES).
Keywords
Heparin-binding growth factors, Heparan sulfate-Proteoglycans, Matrix-remodeling enzymes
References
- Knelson EH, Nee JC, Blobe GC (2014) Heparan sulfate signaling in cancer. Trends Biochem Sci 39: 277–288.
- Sasisekharan R, Venkataraman G (2000) Heparin and heparan sulfate: Biosynthesis, structure and function. Curr Opin Chem Biol 4: 626–631.
- Ishihara M, Takano R, Kanda T, Hayashi K, Hara S, et al. (1995) Importance of 6-O-sulfate groups of glucosamine residues in heparin for activation of FGF-1 and FGF-2. J Biochem 118(6): 1255–60. [Crossref]
- Merry CLR, Lyon M, Deakin J A, Hopwood JJ, Gallagher JT (1999) Highly sensitive sequencing of the sulfated domains of heparan sulfate. J Biol Chem 274: 18455–18462. [Crossref]
- Szatmári T, Dobra K (2013) The role of syndecan-1 in cellular signaling and its effects on heparan sulfate biosynthesis in mesenchymal tumors. Front Oncol 3: 310.
- Farach-carson MC, Carson DD (2007) Perlecan — a multifunctional extracellular proteoglycan scaffold. Glycobiology 17: 897–905. [Crossref]
- Iozzo R V (1994) Perlecan: A gem of a proteoglycan. Matrix Biol 14: 203–208.
- Raman R, Thomas RG, Weiner MW (2010) Border Patrol: Insights into the Unique Role of Perlecan/Heparan Sulfate Proteoglycan2 at Cell and Tissue Borders. 23: 333–336.
- Hammond E, Khurana A, Shridhar V, Dredge K (2014) The Role of Heparanase and Sulfatases in the Modification of Heparan Sulfate Proteoglycans within the Tumor Microenvironment. Front Oncol 4: 1–15. [Crossref]
- Suhovskih A V, Domanitskaya N V, Tsidulko AY, et al. (2015) Tissue-specificity of heparan sulfate biosynthetic machinery in cancer. Cell Adh Migr 9: 452–459. [Crossref]
- Flier JS, Underhill LH, Dvorak HF (1986) Tumors: Wounds That Do Not Heal. N Engl J Med 315: 1650–1659.
- Vreys V, David G (2007) Mammalian heparanase: What is the message? J Cell Mol Med 11: 427–452. [Crossref]
- Uchimura K, Morimoto-Tomita M, Bistrup A, et al. (2006) HSulf-2, an extracellular endoglucosamine-6-sulfatase, selectively mobilizes heparin-bound growth factors and chemokines: effects on VEGF, FGF-1, and SDF-1. BMC Biochem 7: 2. [Crossref]
- Hossain MM, Hosono-Fukao T, Tang R, et al. (2009) Direct detection of HSulf-1 and HSulf-2 activities on extracellular heparan sulfate and their inhibition by PI-88. Glycobiology 20: 175–186. [Crossref]
- Tang R, Rosen SD (2009) Functional consequences of the subdomain organization of the sulfs. J Biol Chem 284: 21505–21514. [Crossref]
- El Masri R, Seffouh A, Lortat-Jacob H, Vivès RR (2017) The “in and out” of glucosamine 6-O-sulfation: the 6th sense of heparan sulfate. Glycoconj J 34: 285–298. [Crossref]
- Ai X, Do AT, Lozynska O, et al. (2003) QSulf1 remodels the 6-O sulfation states of cell surface heparan sulfate proteoglycans to promote Wnt signaling. J Cell Biol 162: 341–351. [Crossref]
- Fellgett SW, Maguire RJ, Pownall ME (2015) Sulf1 has ligand-dependent effects on canonical and non-canonical Wnt signalling. J Cell Sci 128: 1408–1421. [Crossref]
- Tan KW, Chong SZ, Wong FHS, et al. (2013) Neutrophils contribute to inflammatory lymphangiogenesis by increasing VEGF-A bioavailability and secreting VEGF-D. Blood 122: 3666–77.
- Sanderson RD, Yang Y, Kelly T, MacLeod V, Dai Y, et al. (2005) Enzymatic remodeling of heparan sulfate proteoglycans within the tumor microenvironment: Growth regulation and the prospect of new cancer therapies. J Cell Biochem 96: 897–905. [Crossref]
- Kano MR, Morishita Y, Iwata C, Iwasaka S, Watabe T, et al. (2005) VEGF-A and FGF-2 synergistically promote neoangiogenesis through enhancement of endogenous PDGF-B-PDGFRbeta signaling. J Cell Sci 118: 3759–3768. [Crossref]
- Robinson CJ, Mulloy B, Gallagher JT, Stringer SE (2006) VEGF165-binding sites within heparan sulfate encompass two highly sulfated domains and can be liberated by K5 lyase. J Biol Chem 281: 1731–1740. [Crossref]
- Michael Elkin, Neta Ilan, Rivka Ishai-Michaeli, Yael Friedmann, Orit Papo, et al. (2001) Heparanase as mediator of angiogenesis: mode of action. FASEB J 15: 1661–1663.
- Myler HA, West JL (2002) Heparanase and platelet factor-4 induce smooth muscle cell proliferation and migration via bFGF release from the ECM. J Biochem 131: 913–922. [Crossref]
- Reiland J, Kempf D, Roy M, Denkins Y, Marchetti D (2006) FGF2 Binding, Signaling, and Angiogenesis Are Modulated by Heparanase in Metastatic Melanoma Cells. Neoplasia 8: 596–606. [Crossref]
- Duchesne L, Octeau V, Bearon RN, Beckett A, Prior IA, et al. (2012) Transport of fibroblast growth factor 2 in the pericellular matrix is controlled by the spatial distribution of its binding sites in heparan sulfate. PLoS Biol 10: 16. [Crossref]
- Hulett MD, Freeman C, Hamdorf BJ, Baker RT, Harris MJ, et al. (1999) Cloning of mammalian heparanase, an important enzyme in tumor invasion and metastasis. Nat Med 5: 803–809. [Crossref]
- Parish CR, Freeman C, Brown KJ, Francis DJ, Cowden WB (1999) Identification of sulfated oligosaccharide-based inhibitors of tumor growth and metastasis using novel in vitro assays for angiogenesis and heparanase activity. Cancer Res 59: 3433–3441. [Crossref]
- Sikora AS, Hellec C, Carpentier M, Martinez P, Delos M, et al. (2016) Tumour-necrosis factor-α induces heparan sulfate 6-O-endosulfatase 1 (Sulf-1) expression in fibroblasts. Int J Biochem Cell Biol 80: 57–65. [Crossref]
- Langsdorf A, Schumacher V, Shi X, Tran T, Zaia J, et al. (2011) Expression regulation and function of heparan sulfate 6-O-endosulfatases in the spermatogonial stem cell niche. Glycobiology 21: 152–161. [Crossref]
- Chau BN, Diaz RL, Saunders MA, Cheng C, Chang AN, et al. (2009) Identification of SULF2 as a novel transcriptional target of p53 by use of integrated genomic analyses. Cancer Res 69: 1368–1374. [Crossref]
- Holmes RS (2017) Comparative and Evolutionary Studies of Vertebrate Extracellular Sulfatase Genes and Proteins: SULF1 and SULF2. J Proteomics Bioinform 10: 32–40.
- Lai JP, Sandhu DS, Shire AM, Roberts LR (2008) The tumor suppressor function of human sulfatase 1 (SULF1) in carcinogenesis. J Gastrointest Cancer 39: 149–158. [Crossref]
- Bret C, Moreaux J, Schved JF, Hose D, Klein B (2011) SULFs in human neoplasia: Implication as progression and prognosis factors. J Transl Med 9: 72. [Crossref]
- Narita K, Staub J, Chien J, Meyer K, Bauer M, et al. (2006) HSulf-1 inhibits angiogenesis and tumorigenesis in vivo. Cancer Res 66: 6025–6032. [Crossref]
- Lai JP, Chien J, Strome SE, Staub J, Montoya DP, et al. (2004) HSulf-1 modulates HGF-mediated tumor cell invasion and signaling in head and neck squamous carcinoma. Oncogene 23: 1439–1447. [Crossref]
- Lai J, Chien J, Staub J, Avula R, Greene EL, et al. (2003) Loss of HSulf-1 up-regulates heparin-binding growth factor signaling in cancer. J Biol Chem 278: 23107–23117. [Crossref]
- Mondal S, Roy D, Camacho-Pereira J, Khurana A, Chini E, et al. (2015) HSulf-1 deficiency dictates a metabolic reprograming of glycolysis and TCA cycle in ovarian cancer. Oncotarget 6: 33705–33719. [Crossref]
- Khurana A, Beleford D, He X, Chien J, Shridhar V (2013) Role of heparan sulfatases in ovarian and breast cancer. Am J Cancer Res 3: 34–45. [Crossref]
- Lee HY, Yeh BW, Chan TC, Yang KF, Li WM, et al. (2017) Sulfatase-1 overexpression indicates poor prognosis in urothelial carcinoma of the urinary bladder and upper tract. Oncotarget 8: 47216–47229. [Crossref]
- Hao NB, Lü MH, Fan YH, Cao YL, Zhang ZR, et al. (2012) Macrophages in tumor microenvironments and the progression of tumors. Clin Dev Immunol 2012: 948098. [Crossref]
- Solinas G, Schiarea S, Liguori M, Fabbri M, Pesce S, et al. (2010) Tumor-conditioned macrophages secrete migration-stimulating factor: a new marker for M2-polarization, influencing tumor cell motility. J Immunol 185: 642–652. [Crossref]
- Balkwill FR, Mantovani A (2012) Cancer-related inflammation: Common themes and therapeutic opportunities. Semin Cancer Biol 22: 33–40. [Crossref]
- Solinas G, Germano G, Mantovani A, Allavena P (2009) Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol 86: 1065–1073. [Crossref]
- Germano G, Allavena P, Mantovani A (2008) Cytokines as a key component of cancer-related inflammation. Cytokine 43: 374–379. [Crossref]
- Silzle T, Kreutz M, Dobler MA, Brockhoff G, Knuechel R, et al. (2003) Tumor-associated fibroblasts recruit blood monocytes into tumor tissue. Eur J Immunol 33: 1311–1320. [Crossref]
- Li R, Hebert JD, Lee TA, Xing H, Boussommier-Calleja A, et al. (2017) Macrophage-secreted TNFα and TGFβ1 Influence Migration Speed and Persistence of Cancer Cells in 3D Tissue Culture via Independent Pathways. Cancer Res 77: 279–290. [Crossref]
- Barron DA, Rowley DR (2012) The reactive stroma microenvironment and prostate cancer progression. Endocr Relat Cancer 19: 187–204. [Crossref]
- Miao HQ, Liu H, Navarro E, Kussie P, Zhu Z (2006) Development of Heparanase Inhibitors for Anti-Cancer Therapy. Curr Med Chem 13: 2101–2111. [Crossref]
- McKenzie EA (2007) Heparanase: a target for drug discovery in cancer and inflammation. Br J Pharmacol 151: 1–14. [Crossref]