Abstract
In a century of research, it has gradually become clear that glucagon should no longer be considered only as a counter-regulatory hormone of insulin accordingly to its role in the physiopathogenesis of metabolic pathologies such as diabetes, obesity and fatty liver appears to be decisive. As hyperglucagonemia represents the common feature of various metabolic pathologies not only in adults but also in pediatric patients, glucagon can be a problem but also a solution in the field of metabolic diseases. In fact, opposing therapeutic strategies have been developed which inhibit or enhance the activity of glucagon depending on the clinical situation and are also applied in pediatric age. This review aims to take stock of the situation on the physiopatogenetic role of glucagon in metabolic pathologies and bring together the dots of recent discoveries leading to the hypothesis of new solutions in the management and prevention ofthesepathologies.
Keywords
Glucagon, NAFLD, Obesity, Diabetes, Children
Introduction
In 2023 we celebrated the centenary of the discovery of glucagon, which occurred almost by chance since it was initially isolated as a contaminant of the first insulin preparations in 1923. However, the hormonal role of glucagon was only established in the 1950s. Recently, animal and human studies have confirmed the essential role of glucagon in glucose metabolism but have suggested equal importance for amino acid and lipid metabolism [1]. As considered an anti-insulin hormone, it was early on used to treat insulin-induced hypoglycemic coma episodes in people with Type 1 Diabetes Mellitus (T1DM). Nevertheless, a key step in the history of glucagon has been the discovery of its role and the role of α-cells in the physiology and pathophysiology of Type 2 diabetes (T2DM) and obesity [2]. In the last decades, research on glucagon has been slowed down by the difficulty encountered in carrying out glucagonemia measurements [3] which seems to have been overcome thanks to the development of a new high-quality ELISA method [4]. Currently, a century after the discovery of glucagon, there is still a lot to learn about this second pancreatic hormone and it seems necessary to re-elaborate the discoveries achieved so far to lay the foundations for innovative research projects.
Necessary Physiology Hints
Glucagon was initially known to be antagonist to insulin for its opposite metabolic effects on glucose metabolism. In particular, glucagon acts directly on glucose metabolism through three main mechanisms: in the liver, glucagon increases glucose production by stimulating glycogenolysis and gluconeogenesis [5] while in adipose tissue, glucagon stimulates lipolysis with the release of fatty acids and subsequent formation of ketone bodies in the liver [6] resulting both in a net increase of blood glucose levels; in contrast, glucagon acts on β-cells by inhibiting insulin production, thereby, giving a major contribute in maintaining glucose homeostasis. Therefore, glucagon binds specifically to Glucagon Receptor (GCGR), detected mainly in b-cells, liver cells and adipocytes [7]. However, the glucagon receptor has a wide distribution in the body and this explains its multiple known and potential effects. In fact, GCGR is also found in kidneys, heart, lymphoblasts, spleen, brain, adrenal glands, retina, and gastrointestinal tract [8]. Glucagon also controls indirectly blood sugar levels in the kidney through renal excretion by increasing water reabsorption and glomerular filtration and thereby glucose reabsorbtion [9]. Nevertheless, it is currently known that the role of glucagon is not limited to maintaining glucose homeostasis. In fact, glucagon appears to be the basis of a physiological response of satiety induced by meal, as glucagon concentrations increase during the consumption of a mixed meal [10]. The regulatory mechanisms that control glucagon-induced satiety are poorly understood but mediation of vagal afferent fibers in the hepatic branch that transmit signals to the central nervous system is hypothesized [5]. Furthermore, Glucagon promotes weight loss having a direct effect on slowing gastric emptying and increasing energy expenditure [11]. The mechanism of action of glucagon in the remaining areas of the body where its receptor is represented, such as retina, heart and gatrointestinal tract, still remains to be clarified.
Glucagon and Liver-α cell Axis
The main end organ for glucagon is the liver where a feedback axis, the “liver-alpha cell axis” (Figure 1), has been established [12]. In fact, the net increase in hepatic plasma glucose secretion, due to glucagon induced glycogenolysis and gluconeogenesis, determines a direct inhibition of glucagon secretion from α-cells. Furthermore, glucagon increases hepatic absorption and turnover of amino acids, leading to decreased aminoacids levels and inducing, thereby, ureagenesis, which again reduces the secretion of glucagon. Also, glucagon increases hepatic β-oxidation and decreases lipogenesis, lowering the circulating concentration of free fatty acids (FFAs). Although, a plausible mechanism through which lower circulating FFAs may inhibit glucagon secretion has not yet been established [6].
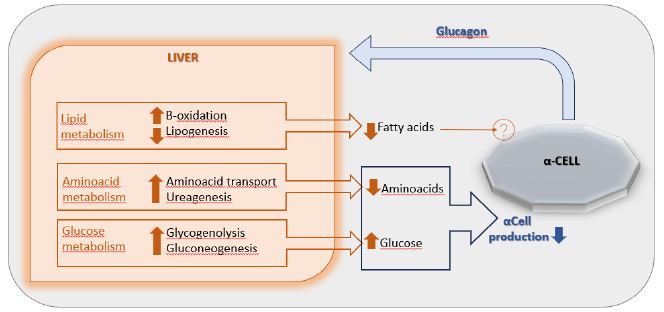
Figure 1: The liver-α-cell-axis in health. Modified from American Diabetes Association [The Liver-α-Cell Axis in Health and in Disease, American Diabetes Association, 2022]. Copyright and all rights reserved. Material from this publication has been used with the permission of American Diabetes Association.
Hyperglucagonemia: The Main Character
Metabolic disorders have long been thought to be caused by total or relative insulin deficiency: this is known as Insulin-centric theory [13]. However, in 1978 Unger and collaborators, in contrast to the insulinocentric theory and in light of discovery of the effects of glucagon, proposed the theory of bihormonal regulation [14]. They found that some metabolic disturbances associated with diabetes, such as elevated lipolysis, increased proteolysis, and impaired glucose utilization are directly caused by insulin deficiency; while others, such as decreased glycogensynthesis, increased ketogenesis, elevated liver glycogenolysis, and gluconeogenesis, are direct effects of excess glucagon. Lately, between the end of the twentieth century and the beginning of the twentyfirst century, the glucagonocentric theory was established, already intuited by Unger and his collaborators, supported by the following evidence: in mice lacking GCGR, insulin deficiency does not cause hyperglicemia, in humans hyperglucagonemia has been established in all forms of diabetes, therefore, excess glucagon represents the sine qua non for the development of hyperglycemia [15]. Physiologically, hypoglycemia represents the main stimulus to glucagon secretion. However, in individuals with diabetes, therefore in conditions of hyperglycemia, there is a paradoxical increase in glucagon in conditions of hyperglycemia. Until recently, this dynamic, which leads to hyperglucagonemia, was explained exclusively through the tonic inhibition exerted by insulin on α-cells, in light of the concept of unidirectional flow from beta to alpha cells [16]. Until the 2000s, it was, therefore, thought that the impact of alpha cells on β cell function was negligible, probably because the studies were mostly based on rodent islets in which α-cells are less represented than in humans [17]. Eventually, in the new millennium, a more sophisticated model of intra-islets vascular system with bidirectional flow and circulation integrated with the exocrine pancreas, was recognized. Therefore, an active role of α-cells has been recognized from both a physiological and pathophysiological point of view, leading to the concept of cross-talk between alpha- and beta-cells [18].
The Role of the Inter-cellular Cross-talk
Glucagon and insulin receptors are expressed on both alpha- and beta-cells, proving that there is a reciprocal relationship between them. Insulin exerts a tonic inhibition on glucagon production by α-cells directly through insulin receptor, therefore, a decrease in insulin induces increased glucagon production [19]. As GCGRs are more abundant in β-cells than Insulin Receptors in α-cells, it has been demonstrated that glucagon secretion acts a direct effect on insulin release [20]. Moreover, in condition of hyperglycemia, β-cells in close contact with the alpha cells release more insulin compared with β-cells deprived of these contacts [21]. It has also been shown that people with T2DM show elevated α-cell-to-β-cell mass ratios, potentially because α-cells are necessary for mantaining β-cell insulin secretion [22]. Although the action on GCGR, glucagon seems to stimulate insulin secretion predominantly via the GLP1 (glucagon-like peptide 1) receptor expressed on β-cell surface [23].
Hyperglucagonemia: The Common Feature
It is known that T1DM and T2DM recognize a different pathogenesis, but these two pathologies have in common hyperglucagonemia whose pathogenetic role has long been overlooked. Lack of postprandial suppression and subsequent glucagon hypersecretion is characteristic in patients with T1DM or T2DM [24]. Even individuals with subtle glucose metabolism disturbances without having clear diabetes mellitus may have excess glucagon in response to the OGTT [25]. Different causes of hyperglucagonemia can be hypothesized and, although it seems difficult to make a clear distinction between metabolic pathologies, since some of them constitute a continuum, recognizing the predominant mechanism in each of them could guide the therapeutic choice and determine a better efficacy, as summarized in the Table 1.
Table 1: Different causes of hyperglucagonemia
|
Main causes of hyperglucagonemia |
Metabolic pathologies |
| 1) Lack of suppression from insulin deficit | T1DM |
| 2) Role of incretins | T2DM – OBESITY |
| 3) Liver glucagon receptor resistance | T2DM – OBESITY |
| 4) altered liver-alpha cell axis | NAFLD |
Hyperglucagonemia in Obesity and NAFLD
It is known that Nonalcoholic Fatty Liver Disease (NAFLD) represents the most common chronic liver disease in children and adolescents and represents an early risk factor for the development of obesity and T2DM [26]. Studies revealed that hyperglucagonemia is more closely related to obesity and fatty liver disease than to diabetes: fasting hyperglucagonemia also occurs in individuals with obesity and normal glucose tolerance [27]. The proposed hypothesis is that NAFLD drives hepatic resistance to glucagon by altering the liver-alpha cell feedback mechanism (Figure 2) and thus resulting in increased circulating levels of aminoacids that stimulate α-cells to secrete glucagon resulting in hyperglucagonemia [28]. In fact, a study conducted in 2020 showed greater glucagon resistance at the level of liver aminoacid turnover in individuals with obesity and NAFLD compared to healthy lean (non-steatotic) individuals [29]. Given its causal role in hyperglucagonemia, plasma glucagon concentration could also be useful for identifying pediatric patients most at risk for NAFLD [30].
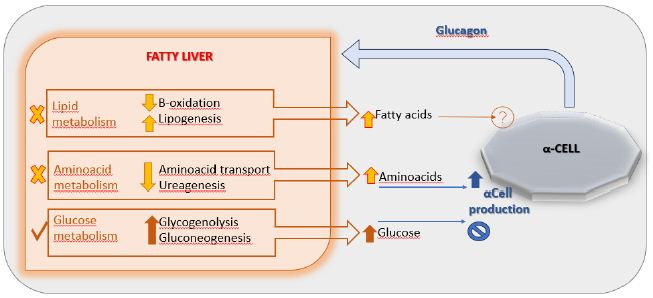
Figure 2: The liver-α-cell-axis in disease. Modified from American Diabetes Association [The Liver-α-Cell Axis in Health and in Disease, American Diabetes Association, 2022]. Copyright and all rights reserved. Material from this publication has been used with the permission of American Diabetes Association.
Hyperglucagonemia in Obesity and T2DM
In metabolic disorders such as T2DM and obesity the alteration of incretin production seems to prevail as possible mechanism responsible for hyperglucagonemia. About that, a study was conducted on patients aged 10 to 18 years with obesity and varying glucose tolerance from Impaired Glucose Tolerance (IGT) up to T2DM compared to controls with normal glucose tolerance. The authors demonstrated that, compared to controls, obese patients with impaired glucose tolerance exhibit a reduction in GLP1 levels in parallel with the increase in postprandial glucagon levels while an increase in fasting glucagon levels in parallel with a reduction in fasting GLP-1 levels [31]. These differences became more evident the more glucose tolerance was reduced. Therefore, an important role must also be recognized in the alteration of incretin levels. In light of this, it seems reasonable to deduce that T2DM therapy with GLP1 has a stronger rationale rather than metformin. Furthermore, a chronic hyperglycemic condition has been shown to increase the expression of the GCGR on the liver and decrease its downstream signaling. This means that a real mechanism of hepatic receptor resistance to glucagon is established [32]. Additionally, it is also hypothesized that the pathophysiology of T2DM is based on a mutation in the gene that codes for the GCGR [33,34].
Hyperglucagonemia and T1DM
In the light of what has been seen on the interaction between alpha- and beta-cells, in subjects affected by T1DM, insulin deficiency leads to the lack of tonic inhibition exerted by β-cell on α-cell, therefore, there is an increase in glucagonemia. Additionally, glucagon seems to play a crucial role especially evident in case of diabetic ketoacidosis (DKA) [35]. Thus, in insulin deficiency, glucagon prevails, FFAs are transferred from the circulation to the mitochondria of the liver cells. Then, the oxidation of FFAs takes place, and acetyl-CoA is produced and is used for the synthesis of ketone bodies [36]. However, there is a difference between the ketogenesis induced by physiological conditions such as fasting in order to find an alternative source of energy and the ketogenesis induced by pathological conditions like uncontrolled T1DM [37] in which it is the result of dysregulated metabolism and a lack of insulin and is not intended to function as an energy source [38]. It is widely known that DKA can cause several adverse events and multiply the risk of developing diabetic complications as ketones lead to increased oxidative stress and inflammation which affect mainly cardiomyocytes, erythrocytes, and endothelial cells [39]. Additionally, elevated plasma ketone concentrations appear to be involved in reducing cell surface insulin receptors, leading to increased Insulin Resistence [40]. Since during DKA glucagon production is increased and is responsible for harmful effects on the body exactly like insulin deficiency, it could probably be useful to intervene on hyperglucagonemia and not just manage hyperglycemia and insulin deficiency.
Therapeutic Perspectives in Metabolic Disorders
Hyperglucagonemia and α-cell hyperplasia drive and accelerate metabolic dysfunction [41]. However, studies indicate that, through intra-island paracrine communication, α-cells could enhance β-cell function and preserve them. In fact, increased secretion of glucagon in metabolic diseases is the result of an α-cell and possibly also gut-derived adaptation for the maintenance of energy balance in favor of the β cells [42]. Whether hyperglucagonemia in metabolic disease is a pathogenic responsible or represents a metabolically helpful adaptation remains unclear [43].
What is the appropriate therapeutic approach? In consideration of the fundamental role that glucagon plays in the pathogenesis of metabolic disorders, the main current therapies and those currently under study are based precisely on the management of glucagonemia. The best choice of type of therapy depends on the type of metabolic disorder and its stage.
Glucagon Antagonism
Hyperglycemia patients treated with insulin is driven, at least in part, by hyperglucagonemia and, therefore, contrastable by antagonization of glucagon secretion or action [44]. GCGR antagonism has been proposed as a pharmacological approach for the treatment of T1DM or T2DM, and it is possible through receptor antagonists, monoclonal antibodies (mAbs) against GCGR and antisense oligonucleotides that reduce receptor expression [45]. GCGR mAbs can also induce b-cell regeneration through the trans-differentiation of a portion of pancreatic α-cells or δ-cells into β-cells [46]. A single dose of REMD-477 (Volagidemab) significantly reduces insulin requirement in patients with T1D improving glycemic control without serious adverse reactions [47]. Data are limited and require further study.
The Multi-effectiveness of GLP-1 Analogues
Last but not least Glp-1 analogues (GLP1A) are now well-known and widely used drugs for the treatment of obesity, but they seem even more effective than insulin and metformin in the management of T2DM and could find application as an additional therapy also in T1DM.
The strength of the GLP1A is represented by its pleiotropy: enhances glucose-dependent insulin secretion; inhibits glucagon secretion; promotes the survival, growth and regeneration of pancreatic β-cells; slows gastric emptying and reduces food intake (GLP1A also find application in the pharmacological therapy of pediatric obesity)[48].
It is reasonable to assume that even with GCGR mutations in β-cells, the binding of glucagon to GLP-1R is conserved, therefore, GLP-1A overcome the limits of GCGR antagonism too.
GLP-1A in T2DM
Currently, first-line therapies for the treatment of T2DM in children over 10 years of age and adolescents, in addition to diet and exercise, include insulin and metformin while GLP-1A as a second line. Nowdays the incidence of juvenile-onset diabetes (JOD) is increasing accordingly the increasing prevalence of obesity in adolescents [49] and it must be considered that, compared with adult-onset T2DM, JOD is associated to: more severe impairment of pancreatic B-cell function, which is further complicated by the increase in insulin resistance associated with obesity and puberty; higher rates of microvascular and macrovascular complications, despite a shorter disease duration than in other types of diabetes; higher treatment failure rate of metformin, which is used as a first-line drug for type 2 diabetes [50]. Therefore, there will likely be an increasing use of GLP-1A prior to the initiation of insulin given their potential benefits on weight and glycemic control but especially the antagonistic action of glucagon. In fact, a study showed that weekly treatment with Dulaglutide was superior to placebo in improving glycemic control over 26 weeks among young people with type 2 diabetes treated with metformin and/or insulin [51].
GLP-1A in T1DM
In T1DM, residual β-cell function is minimal, if not completely absent. Therefore, GLP-1A cannot have any effect on the stimulation of insulin secretion in these subjects. In addition to glycemic control which represents the target of insulin therapy, two other non-negligible aspects in the management of T1DM concern weight gain and the paradoxical increase in glucagon refractory to the action of the administered insulin [52]. A study demonstrated better glycemic control, weight reduction, a lower insulin daily dose and especially a significant reduction in total and postprandial glucagon levels in patients with combined therapy insulin-GLP1A [53]. Moreover, other Authors showed that postprandial glucagon levels tend to progressively increase with the duration of T1DM and correlate positively with deterioration of glycemic control and loss of β-cell function [54]. If GLP-1 levels followed the rising trend of glucagon while GLP-1 is thought to negatively modulate glucagon secretion, there would be a difference between the action obtained from physiological levels of GLP-1 and pharmacological ones during therapy with GLP-1A. In light of these results, a new starting point can be defined for the rationale for the use of GLP-1A in association with insulin therapy. Also, in a recent trial, Liraglutide appears to exert an inhibitory effect on ketogenesis through glucagon reduction [55]. Furthermore, another work shows that Liraglutide, not only markedly suppresses the post-prandial excursion of glucagon in a dose-dependent manner, but it also suppresses fasting plasma FFAs concentrations, and therefore ketogenesis, in patients with T1DM [56].
New Challanges
Hyperglucagonemia represents a fundamental pre-requisite for the development of all forms of diabetes but also obesity, and it is due to insulin deficiency, glucagon receptor resistance, imbalance of incretin secretion, and impaired liver-alpha cell axis. Hepatic steatosis, present in almost all obese pediatric patients, could be the main responsible for the establishment of glucagon-resistance. Therefore, hyperglucagonemia could also be considered a valid marker for the development of metabolic diseases in pediatric patients, as useful tool in the prevention strategy. Whereas, the challenge in pharmacological research is to balance the beneficial effects of glucagon on body weight and lipid metabolism with its hyperglycemic effects. Therefore, dual- and tri-agonists combining glucagon with incretin hormones have been developed and studied as anti-diabetic and anti-obesity therapies [57,58]. The GIP/GLP-1-agonist Tirzepatide has been approved by FDA for the treatment of T2DM, and according to clinical studies, Tirzepatide proved to be more effective than Semaglutide also in reducing body weight in patients with obesity [59]. Finally, among the therapeutic perspectives, the real challenge is to approach metabolic pathologies by trying to broaden the targets of action. What if we were only treating part of diabetes by giving insulin and metformin? What if we also considered glucagon in the management of diabetic ketoacidosis? There are numerous questions still unanswered. Shifting the focus of therapy can represent a winning strategy in the management of metabolic pathologies and this is what we hope for, especially for the pediatric population.
Conflict of Interest Statement
The authors have no conflicts of interest to declare.
Funding Sources
This study was not supported by any sponsor or funder.
Author Contributions
Conceptualization, Writing and Editing – G.D.P. and A.M.;
Project administration and Supervision – F.C.;
All authors read and approved the final manuscript.
References
- Holst JJ (2023) Glucagon 100 years Important, but still enigmatic. Peptides 161: 170942. [crossref]
- Scheen AJ, Lefèbvre PJ (2023) Glucagon, from past to present: a century of intensive research and controversies. Lancet Diabetes Endocrinol 11(2): 129-38. [crossref]
- Holst JJ, Wewer Albrechtsen NJ (2019) Methods and Guidelines for Measurement of Glucagon in Plasma. Int J Mol Sci 20(21): 5416. [crossref]
- Kobayashi M, Maruyama N, Yamamoto Y, Togawa T, et al. (2023) A newly developed glucagon sandwich ELISA is useful for more accurate glucagon evaluation than the currently used sandwich ELISA in subjects with elevated plasma proglucagon‐derived peptide levels. J Diabetes Investig 14(5): 648-58. [crossref]
- Müller TD, Finan B, Clemmensen C, DiMarchi RD, et al. (2017) The New Biology and Pharmacology of Glucagon. Physiol Rev 97(2): 721-66.
- Galsgaard KD, Pedersen J, Knop FK, Holst JJ, et al. (2019) Glucagon Receptor Signaling and Lipid Metabolism. Front Physiol 10: 413.
- Zhang H, Qiao A, Yang D, Yang L, et al. (2017) Structure of the full-length glucagon class B G-protein-coupled receptor. Nature 546(7657): 259-64. [crossref]
- Wendt A, Eliasson L (2020) Pancreatic α-cells – The unsung heroes in islet function. Semin Cell Dev Biol 103: 41-50. [crossref]
- Bankir L, Bouby N, Blondeau B, Crambert G (2016) Glucagon actions on the kidney revisited: possible role in potassium homeostasis. Am J Physiol-Ren Physiol 311(2): F469-86. [crossref]
- Unger RH, Orci L (1976) Physiology and pathophysiology of glucagon. Physiol Rev 56(4): 778-826.
- Heppner KM, Habegger KM, Day J, Pfluger PT, et al. (2010) Glucagon regulation of energy metabolism. Physiol Behav 100(5): 545-8.
- Hædersdal S, Andersen A, Knop FK, Vilsbøll T (2023) Revisiting the role of glucagon in health, diabetes mellitus and other metabolic diseases. Nat Rev Endocrinol 19(6): 321-35. [crossref]
- Banting FG, Best CH, Collip JB, Campbell WR, et al. (1922) Pancreatic Extracts in the Treatment of Diabetes Mellitus. Can Med Assoc J 12(3): 141-6. [crossref]
- Unger RH (1978) Role of glucagon in the pathogenesis of diabetes: The status of the controversy. Metabolism 27(11): 1691-709. [crossref]
- Lee Y, Berglund ED, Wang M, Fu X, et al(2012) Metabolic manifestations of insulin deficiency do not occur without glucagon action. Proc Natl Acad Sci 109(37): 14972-6.
- Samols E, Stagner JI, Ewart RB, Marks V(1988) The order of islet microvascular cellular perfusion is B—-A—-D in the perfused rat pancreas. J Clin Invest 82(1): 350-3. [crossref]
- Cabrera O, Berman DM, Kenyon NS, Ricordi C, et al. (2006) The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc Natl Acad Sci 103(7): 2334-9. [crossref]
- Almaça J, Caicedo A (2020) Blood Flow in the Pancreatic Islet: Not so Isolated Anymore. Diabetes 69(7): 1336-8. [crossref]
- Ishihara H, Maechler P, Gjinovci A, Herrera P-L, et al. (2003) Islet β-cell secretion determines glucagon release from neighbouring α-cells. Nat Cell Biol 5(4): 330-5. [crossref]
- Habegger KM, Heppner KM, Geary N, Bartness TJ, et al. (2010) The metabolic actions of glucagon revisited. Nat Rev Endocrinol 6(12): 689-97. [crossref]
- Wojtusciszyn A, Armanet M, Morel P, Berney T, et al. (2008) Insulin secretion from human beta cells is heterogeneous and dependent on cell-to-cell contacts. Diabetologia 51(10): 1843-52. [crossref]
- Fujita Y, Kozawa J, Iwahashi H, Yoneda S, et al. (2018) Human pancreatic α‐ to β‐cell area ratio increases after type 2 diabetes onset. J Diabetes Investig 9(6): 1270-82. [crossref]
- Svendsen B, Larsen O, Gabe MBN, Christiansen CB, et al. (2018) Insulin Secretion Depends on Intra-islet Glucagon Signaling. Cell Rep 25(5): 1127-1134.e2. [crossref]
- Brown RJ, Sinaii N, Rother KI (2008) Too Much Glucagon, Too Little Insulin. Diabetes Care 31(7): 1403-4.
- Bagger JI, Knop FK, Lund A, Holst JJ, Vilsbøll T(2014) Glucagon responses to increasing oral loads of glucose and corresponding isoglycaemic intravenous glucose infusions in patients with type 2 diabetes and healthy individuals. Diabetologia 57(8): 1720-5. [crossref]
- Smith SK, Perito ER (2018) Nonalcoholic Liver Disease in Children and Adolescents. Clin Liver Dis 22(4): 723-33.
- Wewer Albrechtsen NJ, Junker AE, Christensen M, Hædersdal S, et al. (2018) Hyperglucagonemia correlates with plasma levels of non-branched-chain amino acids in patients with liver disease independent of type 2 diabetes. Am J Physiol-Gastrointest Liver Physiol 314(1): G91-6. [crossref]
- Suppli MP, Lund A, Bagger JI, Vilsbøll T, et al. (2016) Involvement of steatosis-induced glucagon resistance in hyperglucagonaemia. Med Hypotheses 86: 100-3. [crossref]
- Suppli MP, Bagger JI, Lund A, Demant M, Van Hall G, Strandberg C, et al(2020) Glucagon Resistance at the Level of Amino Acid Turnover in Obese Subjects With Hepatic Steatosis. Diabetes 69(6): 1090-9. [crossref]
- Castillo‐Leon E, Cioffi CE, Vos MB(2020) Perspectives on youth‐onset nonalcoholic fatty liver disease. Endocrinol Diabetes Metab 3(4): e00184.
- Manell H, Staaf J, Manukyan L, Kristinsson H, et al. (2016) Altered Plasma Levels of Glucagon, GLP-1 and Glicentin During OGTT in Adolescents With Obesity and Type 2 Diabetes. J Clin Endocrinol Metab 101(3): 1181-9. [crossref]
- Bozadjieva Kramer N, Lubaczeuski C, Blandino-Rosano M, Barker G, et al. (2021) Glucagon Resistance and Decreased Susceptibility to Diabetes in a Model of Chronic Hyperglucagonemia. Diabetes 70(2): 477-91. [crossref]
- Hager J, Hansen L, Vaisse C, Vionnet N, et al(1995) A missense mutation in the glucagon receptor gene is associated with non-insulin-dependent diabetes mellitus. Nat Genet 9(3): 299-304.
- Gough SCL, Saker PJ, Pritchard LE, Merriman TR, et al. (1995) Mutation of the glucagon receptor gene and diabetes mellitus in the UK: association or founder effect? Hum Mol Genet 4(9): 1609-12.
- Veneti S, Grammatikopoulou MG, Kintiraki E, Mintziori G, et al. (2023) Ketone Bodies in Diabetes Mellitus: Friend or Foe? Nutrients 15(20): 4383.
- Shi L, Tu BP (2015) Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr Opin Cell Biol 33: 125-31.
- Sherwin RS, Hendler RG, Felig P(1976) Effect of Diabetes Mellitus and Insulin on the Turnover and Metabolic Response to Ketones in Man. Diabetes 25(9): 776-84. [crossref]
- Hall S, Wastney M, Bolton T, Braaten J, et al. (1984) Ketone body kinetics in humans: the effects of insulin-dependent diabetes, obesity, and starvation. J Lipid Res 25(11): 1184-94. PMID: 6440941. [crossref]
- Kanikarla-Marie P, Jain SK(2015) Hyperketonemia (Acetoacetate) Upregulates NADPH Oxidase 4 and Elevates Oxidative Stress, ICAM-1, and Monocyte Adhesivity in Endothelial Cells. Cell Physiol Biochem 35(1): 364-73. [crossref]
- Kanikarla-Marie P, Jain SK(2016) Hyperketonemia and ketosis increase the risk of complications in type 1 diabetes. Free Radic Biol Med 95: 268-77.
- Lee YH, Wang M-Y, Yu X-X, Unger RH (2016) Glucagon is the key factor in the development of diabetes. Diabetologia 59(7): 1372-5.
- Zhang Y, Han C, Zhu W, Yang G, et al. (2021) Glucagon Potentiates Insulin Secretion Via β-Cell GCGR at Physiological Concentrations of Glucose. Cells 10(9): 2495. [crossref]
- Finan B, Capozzi ME, Campbell JE (2020) Repositioning Glucagon Action in the Physiology and Pharmacology of Diabetes. Diabetes 69(4): 532-41. [crossref]
- Unger RH, Cherrington AD (2012) Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest 122(1): 4-12. [crossref]
- Patil M, Deshmukh NJ, Patel M, Sangle GV (2020) Glucagon-based therapy: Past, present and future. Peptides 127: 170296. [crossref]
- Gu L, Cui X, Lang S, Wang H, et al. (2019) Glucagon receptor antagonism increases mouse pancreatic δ-cell mass through cell proliferation and duct-derived neogenesis. Biochem Biophys Res Commun 512(4): 864-70. [crossref]
- Pettus J, Boeder SC, Christiansen MP, Denham DS, et al. (2022) Glucagon receptor antagonist volagidemab in type 1 diabetes: a 12-week, randomized, double-blind, phase 2 trial. Nat Med 28(10): 2092-9. [crossref]
- Toft-Nielsen M-B, Damholt MB, Madsbad S, Hilsted LM, et al. (2001) Determinants of the Impaired Secretion of Glucagon-Like Peptide-1 in Type 2 Diabetic Patients. J Clin Endocrinol Metab 86(8): 3717-23. [crossref]
- Pyle L, Kelsey MM (2021) Youth-onset type 2 diabetes: translating epidemiology into clinical trials. Diabetologia 64(8): 1709-16. [crossref]
- TODAY Study Group (2012) A Clinical Trial to Maintain Glycemic Control in Youth with Type 2 Diabetes. N Engl J Med 366(24): 2247-56.
- Arslanian SA, Hannon T, Zeitler P, Chao LC, et al. (2022) Once-Weekly Dulaglutide for the Treatment of Youths with Type 2 Diabetes. N Engl J Med 387(5): 433-43.
- Frandsen CS, Dejgaard TF, Madsbad S (2016) Non-insulin drugs to treat hyperglycaemia in type 1 diabetes mellitus. Lancet Diabetes Endocrinol 4(9): 766-80.
- Ilkowitz JT, Katikaneni R, Cantwell M, Ramchandani N, et al. (2016) Adjuvant Liraglutide and Insulin Versus Insulin Monotherapy in the Closed-Loop System in Type 1 Diabetes: A Randomized Open-Labeled Crossover Design Trial. J Diabetes Sci Technol 10(5): 1108-14. [crossref]
- Fredheim S, Andersen M-LM, Pörksen S, Nielsen LB, et al. (2015) The influence of glucagon on postprandial hyperglycaemia in children 5 years after onset of type 1 diabetes. Diabetologia 58(4): 828-34. [crossref]
- Garg M, Ghanim H, Kuhadiya ND, Green K, et al. (2017) Liraglutide acutely suppresses glucagon, lipolysis and ketogenesis in type 1 diabetes. Diabetes Obes Metab 19(9): 1306-11. [crossref]
- Kuhadiya ND, Dhindsa S, Ghanim H, Mehta A, et al. (2016) Addition of Liraglutide to Insulin in Patients With Type 1 Diabetes: A Randomized Placebo-Controlled Clinical Trial of 12 Weeks. Diabetes Care 39(6): 1027-35. [crossref]
- Urva S, Coskun T, Loh MT, Du Y, et al. (2022) LY3437943, a novel triple GIP, GLP-1, and glucagon receptor agonist in people with type 2 diabetes: a phase 1b, multicentre, double-blind, placebo-controlled, randomised, multiple-ascending dose trial. The Lancet 400(10366): 1869-81. [crossref]
- Knerr PJ, Mowery SA, Douros JD, Premdjee B, et al. (2022) Next generation GLP-1/GIP/glucagon triple agonists normalize body weight in obese mice. Mol Metab 63: 101533. [crossref]
- Heise T, Mari A, DeVries JH, Urva S, Li J, Pratt EJ, et al. (2022) Effects of subcutaneous tirzepatide versus placebo or semaglutide on pancreatic islet function and insulin sensitivity in adults with type 2 diabetes: a multicentre, randomised, double-blind, parallel-arm, phase 1 clinical trial. Lancet Diabetes Endocrinol 10(6): 418-429. [crossref]