Abstract
The paper begins with a review of known diffusion models of the literature, in terms of the mass fraction and the chemical potential. Next the thermodynamic consistency of the pertinent schemes is investigated and it follows that the recourse to the chemical potential is justified in two different ways. The alternative recourse to the balance equations, through a linearization procedure, is suggested as a check of consistency. Finally it is pointed out that, thanks to the correspondence probability-concentration, the classical models of diffusion can be extended to the quantum context.
Keywords
Diffusion models, Thermodynamic consistency, Concentration, Chemical potential
Introduction
The purpose of this paper is to review standard models of diffusion and to clarify the physical bases leading to the different differential equations. To understand the basic starting point and to establish the required notation we start with addressing attention to a mixture of n fluid constituents.
In essence, let ρα , vα be the mass density and the velocity of the α th constituent,
α = 1, …, n. We let
![]()
be the mass density of the mixture and the mass fraction (or concentration) of the α th constituent; the sum on α is understood from 1 to n. As we prove in a moment ωα is subject to the differential equation
![]()
where hα is the mass flux, ∇ is the gradient operator and then ∇ is the divergence, and the superposed dot denotes the (total) time derivative.
Within the theory of mixtures hα is clearly defined and shown to satisfy a differential equation [1,2]. As we emphasize in this paper, hα is regarded as an unknown vector to be determined through mathematical assumptions associated with physical properties of the mixture. The original Fick’s model is based on the assumption
![]()
where Dα is the diffusivity. Next other models and their generalizations have been developed in terms of the chemical potential.
In this paper we first review known diffusion models in terms of the mass fraction ωα and the chemical potential µα. Next we investigate the thermodynamic consistency of the pertinent schemes and find that the recourse to the chemical potential is justified in two different ways. Next we examine the alternative recourse to the balance equations through a linearization procedure. Further, we point out that the ideas about the correspondence probability-concentration is applied to modelling diffusion in the quantum context.
Balance Equations for Fluid Mixtures
Consider a mixture of n fluid constituents (see, e.g., [1]). The suffices α, β = 1, 2, …, n label the quantities related to the α th, β th constituent. Hence ρα is the mass density and vα the velocity of the α th constituent. The continuity equation of the α th constituent comprises the mass supply τα, per unit volume and unit time, so that
![]()
The conservation of mass of the whole mixture implies
![]()
Hereafter ![]() is a shorthand for
is a shorthand for ![]() The mass supply τα is nonzero in chemical reactions and phase transformations or generally whenever a constituent may gain or lose mass in favor of the other constituents.
The mass supply τα is nonzero in chemical reactions and phase transformations or generally whenever a constituent may gain or lose mass in favor of the other constituents.
The mass density ρ and the (barycentric) velocity v of the mixture are defined by
![]()
The ratio
![]()
is the mass fraction (or concentration) of the α th constituent and uα = vα − v
is the diffusion velocity. The definition of v implies that
![]()
The sum of (1) with the constraint (2) results in
∂t ρ + ∇ · (ρv) = 0, (3)
which is the continuity equation for the whole mixture.
We now investigate the evolution equation for ωα. Replace ρα with ρωα and vα with
v + uα in (1). In view of (3) we find
ρ(∂tωα + v · ∇ωα) + ∇ · (ραuα) = τα.
Observe that ∂tωα + v. ∇ωα is the derivative with respect to the barycentric observer. As with any function it is denoted by a superposed dot. The vector
hα:= ρα uα
is the α th diffusion flux representing the flux of the α th constituent relative to the barycentric observer. Hence we can write
![]()
The mass fraction ωα, in the barycentric reference, evolves according to (4). This equation is operative once τα and hα are given in terms of ![]() , possibly parameterized by temperature and pressure.
, possibly parameterized by temperature and pressure.
The equation of motion of the α th constituent can be written in the form
∂t (ραvα) + ∇· (ραvα ⊗ vα) − ∇ · Tα − ραbα = mα,
where Tα is the Cauchy stress tensor, bα is the body force, and mα is the growth of the linear momentum, that is the force on the α th constituent due to other constituents of the mixture. The growths {mα} are subject to the constraint
![]()
Modelling of Diffusion Fluxes and Diffusion Equations
The physical modelling of diffusion is most often restricted to non-reacting mixtures, τα = 0, and is based on the view that diffusion is governed by (4) with appropriate models for hα.
The simplest and best known model of diffusion traces back to Fick [3] and is based on an assumption on hα that is motivated by the analogy with the Fourier model of heat conduction. First the total derivative ![]() is replaced with the partial derivative ∂t ωα; this means that diffusion is examined in the barycentric frame. Moreover, Fick’s law assumes the diffusion flux hα is antiparallel to ωα, i.e. hα = κα ωα, where κα > 0 may depend on the constituent. Hence divide eq. (4) by ρ to obtain
is replaced with the partial derivative ∂t ωα; this means that diffusion is examined in the barycentric frame. Moreover, Fick’s law assumes the diffusion flux hα is antiparallel to ωα, i.e. hα = κα ωα, where κα > 0 may depend on the constituent. Hence divide eq. (4) by ρ to obtain
![]()
where ζα = τα /ρ. If κα is constant then the evolution equation for ωα takes the form
![]()
the quantity Dα = κα /ρ is called diffusivity.
Diffusion in solids is often modelled by letting ([4], ch. 17) hα = −Dα ∇ Nα,
where Nα is the concentration in the form
![]()
![]() being the number of atoms per unit volume.
being the number of atoms per unit volume.
Other diffusion equations are based on the following two main assumptions. Let ψα be the α th Helmholtz free energy and define
![]()
The assumption is hα = −κα ∇ µα.
For definiteness we consider ψα in the form [5]
![]()
If λα is constant then it follows
![]()
and eq. (4) takes the form
![]()
Equation (8) is usually referred to as the Cahn-Hilliard equation [6,7]. It is a fourth-order partial differential equation. If the dependence on ∇ ωα is ignored then the Cahn-Hilliard equation reduces to the second-order parabolic equation (6).
Alternatively it is assumed that the evolution of ωα is in fact a relaxation toward equilibrium governed by [8]
![]()
Hence, letting ψα be given again by the Landau-Ginzburg function (7) we obtain
![]()
Equation (9) is a second-order partial differential equation when ψα depends on ∇ωα ; it is referred to as the Ginzburg-Landau equation or the Allen-Cahn equation. While eq. (6) is based on Fick’s law hα = −κα ∇ ωα , eqs (8) and (9) are based on the assumptions
![]()
The function ![]() is often regarded as the chemical potential. As we see in a moment
is often regarded as the chemical potential. As we see in a moment ![]() differs from the standard (correct) expression of the chemical potential. Moreover even simple thermodynamic considerations would justify the use of the chemical potential.
differs from the standard (correct) expression of the chemical potential. Moreover even simple thermodynamic considerations would justify the use of the chemical potential.
Thermodynamic Test of (10)
The balances of energy and entropy are stated by considering the mixture as a whole body. The balance of energy is taken in the form
![]()
where ε is the internal energy density, per unit mass, D is the stretching tensor, q is the heat flux vector, and r is the external heat supply. Let θ > 0 be the absolute temperature. The second law of thermodynamics is stated as follows: the entropy density η and the entropy flux j satisfy the inequality
![]()
For every admissible process that is every set of constitutive functions compatible with the balance equtaions.
For formal conveneince let
![]()
k being the extra-entropy flux to be determined. Substituting ρr-∇.q from eq. (11)
![]()
Let ψ = ε-θη be the Helmholtz free energy of the mixture. The second law inequality can be written as
![]()
To determine thermodynamic consequences of the second law we now consider
![]()
as the set of independent variables. Moreover we specialize T in the form
![]()
with T = O(D) as D→0. Compute ψ˙ and substitute in the Clausius- Duhem inequality (12). The arbitrariness and linearity of ω¨α , ρ¨, D¨ , θ¨ reduces the possible dependencies of ψ
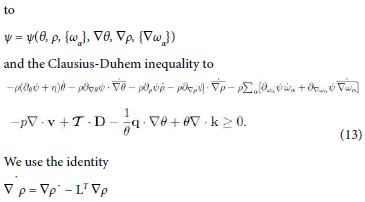
and the analogue for ∇ θ and ∇ ωα. Now, divide (13) by θ and consider the identities
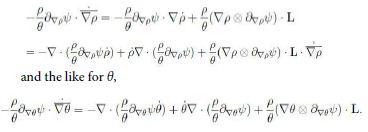
Moreover we have
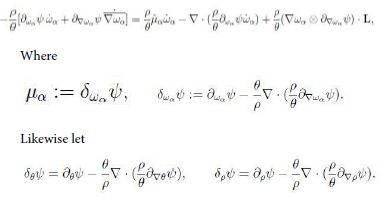
Hence upon some rearrangements we can write inequality (13) in the form
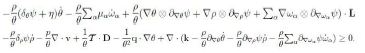
Since L = D+W, with W ∈ skw the spin tensor, then the arbitrariness of W implies that
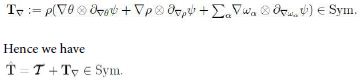
Now, D = ![]() D0 being the deviator of D. This in turn implies that T∇ embodies an isotropic part to contribute to the pressure. For simplicity we let p be free of tr T∇ . Consequently, since ρ˙ = −ρ∇ · v it follows that the inequality takes the form
D0 being the deviator of D. This in turn implies that T∇ embodies an isotropic part to contribute to the pressure. For simplicity we let p be free of tr T∇ . Consequently, since ρ˙ = −ρ∇ · v it follows that the inequality takes the form
![]()
the dots denoting the other terms of the inequality. Hence we let p = ρ2δpψ.
Consequently we are left with the inequality
![]()
Thermodynamic Restriction on ![]()
Sufficient conditions for inequality (14) are
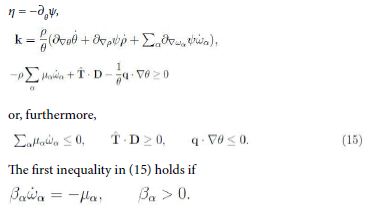
It is suggestive that this thermodynamic condition has the form of (10)2. Yet we have to check whether ![]() Now, from the theory of mixtures we have
Now, from the theory of mixtures we have
![]()
to within kinetic terms. If we let each ψα depend on the corresponding mass density
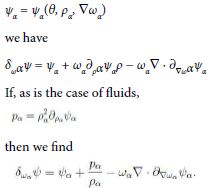
Hence we conclude that, to within ![]() the standard chemical potential is
the standard chemical potential is
![]()
Thermodynamic Restriction on hα
Back to inequality (14) we let again η = −δθ ψ. Next we observe that ![]() is given by (1). Substitution of
is given by (1). Substitution of ![]() from (1) results in
from (1) results in

This inequality holds if each term has the required sign; in particular
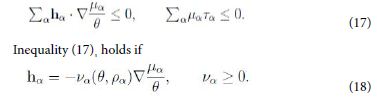
Hence, owing to the neglect of kinetic terms in ψ, by the second law inequality it follows that the diffusion flux hα is determined by the gradient of µα /θ, and not merely of µα. This is a direct thermodynamic requirement without any need of a-priori rescaling [9].
Inequality (17)2 governs the evolution of a possible reaction. For a binary mixture the inequality reduces to
![]()
the reaction proceeds toward the constituent with higher values of the chemical potential.
Diffusion of Electrically Charged Constituents
Equation (4) is a basic reference in the modelling of diffusion.
Indeed, for non-reacting mixtures the basic equation is written in the form
∂tωα = −∇ · hα , (19)
the occurrence of ρ, though non-constant, being ignored; in the literature the notation is frequently ci and Ji for ωα and hα. For definiteness, look at the motion of a charged constituent in a fluid medium.
The diffusion flux is viewed as the sum of three terms [10]: a diffusion term Dα ∇ ωα as in Fick’s law, an advective term ωαv viewed as the transport of the constituent via the motion of the fluid, the diffusivity times the electric force (electromigration). The advective term may be ignored by selecting a frame at rest with the fluid.
An extensive approach in the literature is based on eq. (18) and on hα = −Dα ∇ ωα
in the simplest model. Borrowing from the properties of the ideal gas we take the chemical potential µα in the form
![]()
to within inessential additive terms independent of ωα. Hence we observe that, at constant temperature,
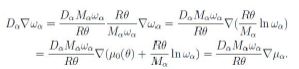
Now the chemical potential is an energy per unit mass. Assume the αth constituent consists of ions with electric charge zαe, where e is (the absolute value of) the electron charge. The force per unit mass is
![]()
φ being the potential and mα the ion mass. Moreover
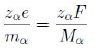
where F is the Faraday constant, i.e. the charge of a mole, and Mα is the molar mass. Hence we write the whole chemical potential, or electrostatic-chemical potential, in the form
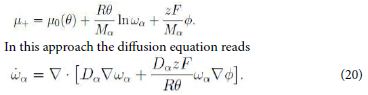
This is a form of the Nernst-Planck equation [11].
Kirkendall Effect
The Kirkendall effect involves a property of diffusion associated with different diffusivities of the constituents. The property was found experimentally by Smiglskas and Kirkendall. In essence, molibdenum markers are located at the boundary between the inner CuZn block and the outer copper covering. Upon heating, the markers are observed to move inward. To explain the experiment, we observe that within the block Cu and Zn atoms have the same density N; since the atomic weights are almost the same (Cu 63.5, Zn 65.4) the equal density N means equal concentration ω. The Zn atoms have a higher diffusivity coefficient and hence the outward flux of Zn is not exactly compensated by the inward flux of Cu atoms. Thus the mass of matter in the block decreases and this results in the movement of the copper- brass interface(markers) toward the inner block.
We now ask for the velocity of the markers. We have
ρZnuZn = hZn = −DZn∇ω, ρCuuCu= hCu = −DCu ∇ω;
really we should account also for a inward flux of vacancies (from the material with the higher diffusion coefficient). In one dimension we obtain the velocity v of the markers as follows,
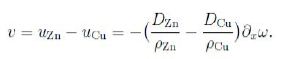
Dynamic Diffusion Equation
Models of diffusion are usually based on the balance equation (4) for the mass fraction ωα and a constitutive equation for the mass flux hα. Hence the resulting differential equation for ωα is strongly affected by the constitutive assumption. It seems of interest to look at the evolution problem via the balance equations for the unknown densities ρα.
Restrict attention to mixtures of inviscid fluids and hence Tα = pα1. Consider the balance of mass and linear momentum in the local form,
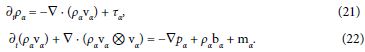
Partial time differentiation of the first equation, divergence of the second one, and substitution of ∇ · ∂t(ραvα) yield
∂2ρα = ∇ · [∇ · (ραvα ⊗ vα)] − ∇ · (∇ · Tα) − ∇· (ραbα) − ∇ · mα + ∂tτα.
If the constituent is regarded as an inviscid fluid, Tα = −pα1, it follows
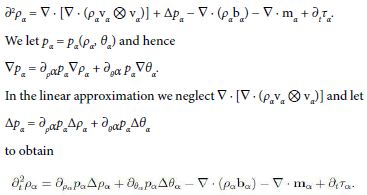
If, further, the temperature is assumed to be uniform, ∇ θα = 0, then
![]()
Equation (23) is the αth equation of a system where ∇. mα and ∂tτα account for possible coupling terms. Since ρα = ρωα, if ρ is assumed to be constant then we have
![]()
If the αth constituent is electrically charged then
![]()
where e is (the absolute value of) the electronic charge, zαe is the ionic charge, and mα
is the ionic mass. In terms of molar quantities we have
![]()
where Mα is the molar mass and F is the Faraday constant, i.e. the charge of a mole. Hence, if E is uniform then we can write the equation for ωα in the form
![]()
Quantum Diffusion Models
If a quantum particle moves in space under a force with potential U then the wavefunction
ψ evolves in time according to the Schr¨odinger equation
![]()
where m is the mass of the particle. Since ψ is complex valued then we can write
![]()
where ρ and S are functions of the position x and time t. The relation
![]()
ascribes to ρ(x, t) the probability density, per unit volume, of finding the particle at the point x at time t. Computing ∂tψ, ∆ψ and substituting in (24) we obtain
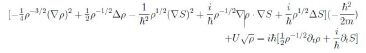
Equating the imaginary parts and observing that ∇ρ · ∇S + ρ∆S = ∇·(ρ∇ S) we have
![]()
This equation has an immediate physical interpretation. If we let ρ be the analogue of the classical mass density then if we let
![]()
we can write (25) in the form
![]()
which is formally the continuity equation in continuum mechanics. This result indicates a close analogy between quantum and classical schemes which are extended to diffusion.
We now look for the possible meaning of the relation coming from equating the real parts. Upon some rearrangements we find
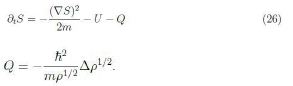
The quantity Q has the role of a potential, like U , and is often referred to as Bohm quantum potential [12]. Furthermore apply the gradient to (26) and assume S has continuous second-order derivatives to have
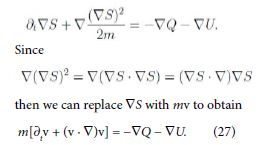
Equation (27) is formally the equation of motion with the Lagrangian (total) time derivative in the left-hand side, as it has to be. Consequently Q may also be viewed as a pressure term.
Diffusion is modelled by having in mind the parabolic character of the classical diffusion equation. Let P be the probability density of Brownian particles with mass m. Form the equation of motion we ascribe to P the equation
![]()
p being the opposite of the pressure. Then we let p = kBθP , where kB is the Boltzmann constant. In the high friction limit [13] it follows the parabolic equation
![]()
When quantum diffusion is described within a continuum mechanics approach the governing equation is taken in a Fick-like form. As an example [14], in the quantum diffusion of H atoms in solid molecular hydrogen films, the H atom concentration, n, is described by a differential equation in the form
![]()
Conclusions
Well-known diffusion equations are considered for the concentration (mass fraction) ωα = ρα/ρ. In addition to the classical parabolic equation (6) we have reviewed the Cahn-Hilliard equation (8) and the Ginzburg-Landau equation (9). The rather general approach to the modelling of diffusion processes is based on the view that the diffusion flux, here hα, has to be determined by a constitutive equation and that a safe rule is to let hα be proportional to the gradient of the chemical potential µα. As a remarkable example we have shown how the approach via chemical potentials, for charged constituents, leads to the Nernst-Planck equation. Diffusion in the quantum domain is still based on the classical Fick’s law.
While hα in terms of ωα is inherited from the historical Fick’s model, the recourse to the chemical potential is found to be thermodynamically consistent in two cases, just those leading to the Cahn-Hilliard and Ginzburg-Landau equations. In the present thermodynamic analysis the αth chemical potential is derived from the Helmholtz free energy
![]()
instead of from ψα(ωα).
The developments of this paper show that the constitutive equations for the mass flux hα are in fact approximations; the involved equation for hα [1] justifies the recourse to approximations. However, the investigation of approximated diffusion equations indicates that a privileged role should be ascribed to the dynamic differential equations, based on the (exact) balance equations (21), (22). The difference in having recourse to the dynamic equations is exemplified with the analogue of Nernst-Planck equation.
Acknowledgments
This research has been developed under the auspices of INDAMCNR.
References
- Müller I (1975). Thermodynamics of mixtures of J. M´ecan. 14: 267-303.
- Morro A (2014). Balance and constitutive equations for diffusion in mixtures of fluids, Meccanica 49: 2109-2123.
- Fick A (1855). Ueber Ann. Phys. 170: 59-86.
- Kittel C (1976). Introduction to Solid State Wiley.
- Landau LD, Khalatnikov IM (1965). On the theory of Oxford.
- Cahn JW (1961). Spinodal Acta Metall. 9: 795-801.
- Cahn JW, Hilliard JE (1971). Spinodal decomposition: a Acta Metall. 19: 151- 161.
- Gurtin ME (1996). Generalized Ginzburg-Landau an Cahn-Hilliard equations based on a microforce Physica D. 92: 178-192.
- Brokate M, Sprekels J (1996). Hysteresis and Phase Springer.
- Kirby BJ (2010). Micro- and Nabnoscale Fluid Mechanics: Transport in Microfluidic Devices, Cambridge University Press.
- Bockris JO’M, Reddy AKN (1970) Modern Plenum Press.
- Bohm D (1952). A suggested interpretation of the quantum theory in terms of “hidden” I. Phys. Rev. 85: 166-179 (1952).
- Tsekov R (2009). Thermo-quantum Int. J. Theor. Phys. 48: 630-636.
- Sheludiakov S, Lee DM, Khmelenko VV, J¨arvinen J, Ahokas J et al. (2021) Purely spatial quantum diffusion of H atoms in solid H2 at temperatures below 1 K, Phys. Rev. Lett. 126: 195301.