DOI: 10.31038/CST.2023824
Simple Summary
The nucleotide excision repair (NER) pathway involves more than thirty protein-protein interactions and removes DNA adducts caused by chemotherapy drugs. The key genes of NER are often over-expressed in cancer cells and alterations of this pathway are responsible for increased or decreased sensitivity to specific therapeutic agents. This is particularly relevant in soft tissue sarcomas (STS), rare mesenchymal-originated tumors whose underlying mechanisms still lack understanding. Altogether the NER pathway components can be potential therapeutic targets in STS. The subtle regulation of NER activity may be clinically relevant as a surrogate prognostic marker or to predict sensitivity to chemotherapy agents. Further prospective evaluation of NER should be performed to address this question.
Abstract
Soft tissue sarcomas (STS) are low-incidence, mesenchymal-derived tumors represented by more than 50 his to types. Despite the latest developments, the rates of patients developing recurrent and metastatic disease are high. Many of the mechanisms underlying STS are still unknown, but there is evidence of the possible role of DNA damage response (DDR) pathways. DDR pathways include a variety of pathways used by cells to repair DNA damage of various kinds; they also have roles in protecting cancer cells from exogenous agents that target DNA, for these reasons are one of the main targets of potential anticancer therapeutic strategies. Nucleotide excision repair (NER) is one of the key repair pathways that can remove various bulky DNA lesions, often given by UV light, and is the main repair mechanism of DNA damage caused by carcinogens and chemotherapeutic drugs. Defects in NER are often the cause of several autosomal recessive genetic diseases. Variations in NER pathway actors can lead to a NER proficiency or NER deficiency condition, and this can be a risk, prognostic, and treatment response factor in cancer. This review focuses on the association between variations in the NER pathway in STS and is intended to point to NER as a pathway to focus on in the next future to optimize the treatments in use and improve the possibilities of personalizing therapies in STS patients in clinical practice.
Keywords
DNA damage response (DDR) mechanism, Nucleotide excision repair (NER), Soft tissue sarcoma (STS), Chemosensitivity
Introduction
Soft tissue sarcomas (STS) are rare tumors with more than 100 different histological subtypes. The scientific community has focused over the years on the search for biomarkers in STS [1] and the identification of variations at the genomic [2], expression [3], and protein [4] level.
Maintaining the integrity of genetic material is critical for the survival of all cell lines, yet various factors, both endogenous and exogenous, can compromise DNA stability. DNA damage repair (DDR) mechanisms are necessary for the maintenance of genome integrity. This is particularly important in cancer cells, in which mechanisms of resistance and sensitivity to radio- and chemotherapeutic cytotoxic agents are directly controlled by DDR pathways. For these reasons, DDR pathways are one of the main targets of potential anticancer therapeutic strategies [5]. The main DDR pathways are direct repair (DR), base excision repair (BER), mismatch repair (MMR), nucleotide excision repair (NER), non-homologous end joining (NHEJ), and homologous recombination repair (HRR) [6]. The absence or deficiency of a specific DDR mechanism can result in genomic instability and tumor progression. Certain modifications of specific genes of a DDR pathway are typical, with some frequency, of some specific cancers [7]. Transcriptomic profiling of tumor tissues suggested codependences between DDR pathways, indicating a potential benefit of combination therapies, which were confirmed by in vitro studies. Somatic alterations in the NER pathway, especially in ERCC genes, are common in various types of cancer. In a 2007 study, Castro et al showed that in cancer cells, NER and apoptosis pathways are the most impaired, with a high diversity of gene expression profiles in comparison to normal cells [8]. Other studies have shown how a deficiency in the NER pathway correlates with increased sensitivity to irofulven and cisplatin [9,10] and decreased sensitivity to trabectedin [11]. Moreover, inhibiting NER has been shown to increase sensitivity to alkylating agents in multiple myeloma cases [12]. DDR pathway alterations are present in numerous histologic subtypes of sarcoma. In a study conducted on STS specimens, at least one pathogenic mutation of the DDR pathway was detected in 15.9% of the patients, the most altered gene was ATRX (10%) and furthermore mutations were observed in 25 sarcoma subtypes. [13]. Recent studies have analyzed the genotype and expression profile of NER genes in STS patients, showing a correlation between ERCC1 and ERCC2 specific single nucleotide polymorphisms (SNPs) and a higher expression of both genes [14].
Nucleotide Excision Repair (NER) Pathway
NER mechanism recognizes and repairs various types of DNA damage caused by UV irradiation, cisplatin, and other damaging agents. NER pathways can be classified as either global genome repair (GGR), which repairs DNA damage anywhere in the genome, or transcription-coupled repair (TCR), which specifically restores DNA strands that are being transcribed (Figure 1). NER mechanisms rely on a series of reactions: recognition of DNA damage, unwinding double-strand DNA in the neighborhood of the damage, excision of the damaged nucleotides, and filling of the single-stranded gap by DNA synthesis. In GGR, DNA damage is recognized by XPC/Rad23 (xeroderma pigmentosum, C/Rad23 complementation group) or UV-DDB (UV-damaged DNA binding protein) [15] while in TC-NER, DNA damage blocks RNA polymerase II (RNAPII) interacting with CSB (ERCC excision repair 6, chromatin remodeling factor) and CSA (ERCC excision repair 8, subunit of CSA ubiquitin ligase complex)-CSB. After damage recognition, in both pathways, RNA polymerase II H transcription initiation factor (TFIIH) is recruited. It is subsequently recruited to the XPG (ERCC excision repair 5, endonuclease) complex, a single-stranded DNA-specific endonuclease. TFIIH unwinds DNA in the vicinity of damage, XPD (ERCC excision repair 2, helicase subunit of the TFIIH core complex), XPB (ERCC excision repair 3, helicase subunit of the TFIIH core complex) and XPA (DNA damage recognition and repair factor) are in charge of recognizing and verifying the damage. XPA binds to the chemically altered nucleotides in a single strand of DNA and recruits the XPF (ERCC excision repair 4, endonuclease catalytic subunit)-ERCC1 (ERCC excision repair 1, endonuclease non-catalytic subunit) catalytic subunit, which makes a cut on the damaged strand of 5′ to extract the damage. Next, XPG makes a 3′ cut that leads to the excision of a single-stranded DNA fragment containing the damage. Then thanks to PCNA (proliferating cell nuclear antigen) and DNA polymerase δ or ε new DNA is synthesized, finally DNA ligase 1 or 3 seals the DNA [16]. Defects in the NER pathway can be attributed to several inherited human diseases, including xeroderma pigmentosum (XP), an autosomal recessive genetic disease characterized by increased sensitivity to UV radiation [17] (Figure 1).
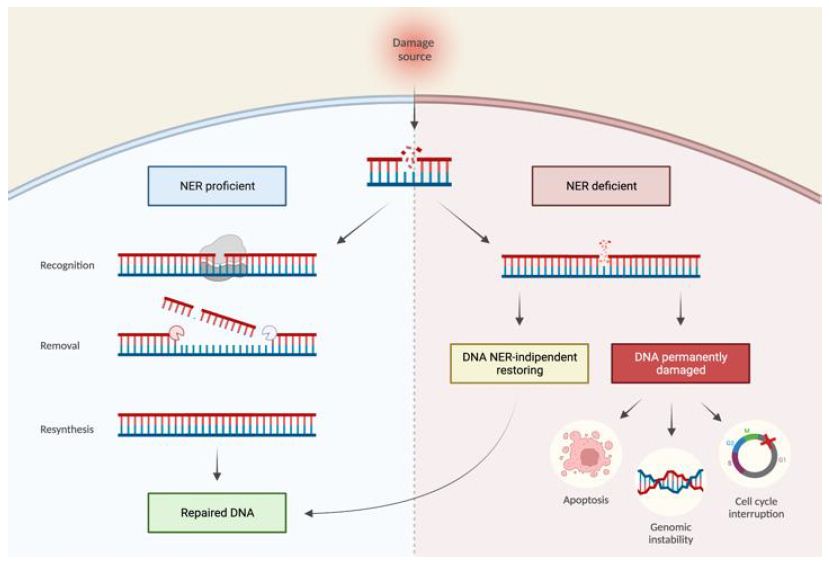
Figure 1: NER proficient and NER deficient tumoral cells. Effects of variations in the NER pathway leading to a NER proficient condition (left) with correct repair of DNA damage or NER deficient (right) with DNA damage persisting. The NER deficient condition can be reversible, resulting in DNA repair, or irreversible, resulting in permanent DNA damage that can lead to cellular damage.
ERCC1
The product of this gene is required for the repair of DNA lesions such as those induced by UV light or formed by electrophilic compounds including cisplatin. The encoded protein forms a heterodimer with the XPF endonuclease, and the heterodimeric endonuclease catalyzes the 5′ incision in the process of excising the DNA lesion. The heterodimeric endonuclease is also involved in HRR and in the repair of inter-strand crosslinks [18]. Overexpression of ERCC1 is correlated with better progression-free survival (PFS) in patients treated with doxorubicin plus trabectedin [19] and favorable overall survival (OS) [19]. Additionally, high expression of ERCC1, and BRCA1 haplotype were associated with the improved progression-free rate (PFR), PFS, and OS in STS [20]. Increased ERCC1 and XPF expression were associated with improved disease-free survival (DFS) and distant disease-free survival (DDFS) in STS [21]. A study carried out on STS patients showed that regarding the SNP rs11615, the alternative allele has a higher germline frequency than the general population and ERCC1 is overexpressed in 75% of STS samples analyzed compared to healthy corresponding tissue and its expression varies according to the genotype [14].
ERCC2 (XPD)
ERCC2 is part of the BTF2/TFIIH complex, which is essential in TCR. The translated protein has ATP-dependent DNA helicase activity and belongs to the RAD3/XPD subfamily of helicases. Defects in this gene are related to Cockayne syndrome, XP cancer-prone complementation group D syndrome, and trichothiodystrophy. [22]. ERCC2 gene is overexpressed in STS and its expression varies according to the genotyping of rs13181 and rs1799793, in addition, these SNPs have a higher frequency than the general population [14]. Additionally, ERCC2 is mutated in 3% of epithelioid sarcoma and 6.5% in perivascular epithelioid cell tumors and is significantly associated with increased homologous recombination deficiency (HRD) scores [13].
ERCC3 (XPB)
This gene encodes an ATP-dependent DNA helicase that is a subunit of basal transcription factor 2 (TFIIH) and, therefore, also functions in class II transcription. Mutations in this gene are associated with XP B, Cockayne’s syndrome, and trichothiodystrophy [23] ERCC3 overexpression is associated with disease progression in STS patients treated with trabectedin [24]. A study analyzing the genetic background, by whole-exome analysis, of a family with a 4-year-old child who has a Li-Fraumeni tumor (often associated with STS) hypothesized that ERCC3 may be a potential TP53-related modifier candidate responsible for accelerated tumor onset by the proband compared with the mother, who carries the same TP53 mutation [25].
ERCC4 (XPF)
XPF forms a complex with ERCC1 by playing a role in the 5′ incision made during NER. This complex is a DNA repair-specific endonuclease that interacts with meiotic structure-specific essential endonuclease 1 (EME1). Variations in this gene can underlie xeroderma pigmentosum complementation group F (XP-F), or xeroderma pigmentosum VI (XP6) [26]. In a study that performed targeted genomic sequencing within an Asian cohort of sarcoma patients, a truncating mutation in ERCC4 (p.Cys723*) was found in two patients with sarcoma diagnosed under 25 years of age [27]. In a retrospective study on angiosarcoma, ERCC4 was found mutated in 6% of patients [28].
ERCC5 (XPG)
This gene encodes a single-stranded DNA-specific endonuclease that makes the 3′ incision in DNA excision repair. XPG also plays a role in RNAPII transcription. Variations in this gene can cause xeroderma pigmentosum complementation group G (XP-G) and Cockayne syndrome [29]. A study of 113 STS samples showed a correlation between high expression of the common allele (aspartic acid at codon 1104) and better PFR, PFS, and OS [20]. A translational study showed that an overexpression of ERCC5 correlates with trabectedin activity and is associated with longer PFS in advanced STS treated with trabectidine [30]. Furthermore, in a cohort of STS, the frequency of SNP rs1047768 is the same as that of the general population, while the frequency of SNP rs2296147 is lower than that of the general population; the gene is overexpressed in 42% of the STSs analyzed and its expression correlates with that of the ERCC2 gene. Finally, the effect of SNP rs1047768 in protein structure was hypothesized for the first time in this study, suggesting a possible effect in ssDNA binding [14]. A meta-analysis associated variations on the ERCC5 gene with an increased risk of STS [31].
ERCC6 (CSB)
The encoded protein has ATP-stimulated ATPase activity, interacts with several transcription and excision repair proteins, and may promote complex formation at DNA repair sites. CSB interacts with RNAPII at the damaged site, and by direct interaction it recruits CSA [32], forming a complex responsible for the association and stabilization of UV-stimulated scaffold protein A (UVSSA), which stimulates TC-NER [33]. Mutations in this gene are associated with Cockayne syndrome type B and cerebro-oculo-facio-skeletal syndrome (COFS) [34]. On dbSNP are reported 68 clinically significant pathogenic variants of ERCC6 [35]. A retrospective translational study on STS showed that ERCC6 was underexpressed in L-sarcomas, compared with other STS subtypes [24].
ERCC8 (CSA)
CSA, encoded by ERCC8 (chr 10), is part of an E3-ubiquitin-ligase complex. CSA, in TC-NER, is required for recovery of DNA synthesis after repair is responsible for ubiquitination and proteasomal degradation of CSB, is required for recovery of DNA synthesis after repair [36] and interacts with CSB and with p44, a subunit of TFIIH. Mutations in this gene have been identified in patients with the hereditary disease of Cockayne syndrome (CS). [34], however genetic polymorphisms are shown to increase breast [37], gastric [38] and oral [39] cancer risk. On dbSNP are reported 32 clinically significant pathogenic variants of ERCC8 [35]. There are no data at present on the correlation of ERCC8 and STS.
XPA
The XPA protein is a zinc finger protein that plays a central role in NER by interacting with DNA and other proteins, forming the structure required to assemble the NER etching complex [40]. A retrospective translational study on STS showed that high levels of XPA expression correlated with better efficacy of trabectedin. [24].
XPC
It is a key component of the XPC complex, which plays an important role in the early stages of GG-NER. It has higher affinity for single-stranded DNA, and is important for damage detection [41]. At present, there are no experimental data on the role of XPC in STS.
TFIIH
TFIIH is a 10-subunit protein complex involved in both transcription and DNA repair, highly conserved in the entire eukaryotic domain. It can be divided in a 7-subunit CORE complex, consisting of XPB, XPD, p62, p44, p34, p52 and p8, and a CAK module (Cyclin Activated Kinase), comprised of CDK7, cyclin H and MAT1 [42]. XPB and XPD are both ATP-dependent DNA helicase and they catalyze the ATP-dependent opening of the DNA at the transcription starting site or at the damaged site. XPB, encoded by ERCC3 in chromosome 2, unwinds the DNA helix in the 3′-5′ direction and can also function as a 5′-3′ DNA traslocase [43], while XPD, encoded by ERCC2 in chromosome 19, acts in 5′-3′ direction and it’s responsible for recruiting the CAK complex [44]. The remaining 5 subunits (p62, p44, p34, p52 and p8) are encoded respectively by GTF2H1 in chromosome 11, GTF2H2 in chromosome 5, GTF2H3 in chromosome 12, GTF2H4 and GTF2H5 in chromosome 6; they carry out structural and ATPase regulation roles [45]. The three-subunit detachable CAK module has a fundamental role as a regulator of both transcription and damage repair pathways; in particular it is necessary for transcriptional activation, but its presence inhibits damage-repair functions [46]. Of the seven genes encoding the CORE components of TFIIH, mutations in ERCC3 and ERCC2 affect both RNA transcription and DNA repair pathway, causing severe disorders such as Xeroderma Pigmentosum, Cockayne Syndrome and Trichothiodystrophy [47]. NCBI dbSNP reports 14 clinically significant pathogenic or likely-pathogenic variants of ERCC3 [35]; in addition, various ERCC3 polymorphisms have been linked to increased risk of lung cancer [48] and osteosarcoma [49]. Regarding ERCC2, dbSNP reports 41 pathogenic or likely-pathogenic variants and polymorphisms in this gene have been associated with a higher risk of lung [50] and colorectal cancer [51]. Furthermore, a significant link has been reported between specific ERCC2 and ERCC3 SNPs and their predisposition to specific types of sarcomas [52]. The other 5 genes of the CORE complex are less affected by clinically significant polymorphisms, although it has been reported that mutations in p52, p8, and p44 are associated with developmental disorders [45]. Regarding the CAK module, high expression of cyclin H has been associated with trabectedin sensitivity in STS [24].
Ubiquitylation in NER Pathway
Ubiquitin is a 76-amino acid protein used for labeling targeted proteins, regulating their stability and function. Ubiquitylation is a sequential process that involves the action of E1 ubiquitin activating enzyme, E2 ubiquitin-conjugating enzyme and E3 ubiquitin ligating enzyme [53]. There are 2 E1, 40 E2 and about 600 E3 enzymes in the human genome [54], which emphasizes the specificity of the ligation process [55]. The initial ubiquitin is attached to the target protein in a lysine (K) residue in the C-terminal portion of the target. Subsequent ubiquitin molecules are sequentially attached to lysine residues of the previous molecule. Poly-ubiquitin chains can have different functions depending on the lysine residue to which the molecules link. The K48-linked chains signal proteasomal degradation of the target protein, whereas K63-linked chains regulate target protein function [56]. Ubiquitylation is reversible by the action of deubiquitinating enzymes (DUBs) [57]. Complex processes such as NER require similarly complex regulation through easily inducible and reversible post-translational modifications. Ubiquitylation has been shown to play a key role in this pathway [58].
GG-NER Regulation by Ubiquitylation
In the presence of UV damage to DNA, the COP9 signalosome is released from the CRL4DDB2 complex, an E3 ubiquitin ligase comprising CUL4, ROC1, and DDB2. In normal conditions the COP9 signalosome inhibits the CRL4DDB2 complex activity, in the absence of this inhibition CUL4 can be neddylated by NEDD8, leading to the activation of the E3 complex [59]. At this stage, recognition of DNA damage by XPC and DDB2 occurs [60]. The CRL4DDB2 complex carries out the action of E3 ubiquitin-ligase on histones H2A, H2B, H3, and H4, weakening the histones-DNA interactions in the damaged area [61]. The complex auto-ubiquitinates DDB2, decreasing its affinity to damaged DNA [62]. This process competes with the presence of XPC at the damaged site, which stabilizes DDB2 [63], and with PARylation of DDB2 by PARP1, which inhibits its ubiquitination [64]. Deubiquitinase BAP1 also appears to be involved in this regulation process [65]. DDB2 is extracted from the complex by VCP/p97 and targeted to the proteasome. The removal of DDB2 from DNA increases the binding affinity of XPC to TFIIH, which is recruited at the damaged site. TFIIH promotes, through its p62 subunit, DDB2 extraction from the complex. XPC-TFIIH-XPA complex formation allows the initiation of the DNA damage verification process [66]. Simultaneously with DDB2 ubiquitylation, the CRL4DDB2 complex also ubiquitinates XPC, not resulting in degradative signaling, but increasing its affinity to DNA [67]. XPC then undergoes SUMOylation, induced by UV damage to DNA, which results in the recruitment of RNF111/Arkadia (SUMO-targeted ubiquitin ligase), responsible for XPC ubiquitylation that leads to its removal from damaged DNA [68]. Extraction of XPC by VCP/p97 allows the other factors of GG-NER to be recruited.
TC-NER Regulation by Ubiquitylation
In the presence of DNA damage, RNA-Pol II is interrupted, recruiting CSA and CSB, which in turn recruit UVSSA. CSA is part of CRL4CSA E3 ubiquitin-ligase complex, comprising CUL4, ROC1 and CSA. As in the GG-NER regulation, this complex is regulated by the COP9 signalosome, which detaches in the presence of DNA damage and allows the neddylation of CUL4, thereby activating the E3 complex [59]. CSB undergoes modification by multiple factors, as it is ubiquitylated by CRL4CSA complex and BRCA1-BARD [69] and deubiquitylated by the deubiquitylating enzyme USP7 [70], which is recruited by UVSSA [71]. This fine-tuned regulation controls the stability of CSB before its extraction by VCP/p97, allowing the recruitment of the other damage repair factors. CRL4CSA complex also mono-ubiquitylates UVSSA, allowing recruitment of TFIIH, which is then linked to RBP1 [60]. If the damage is not repaired, as in the case of mutations in CSA or CSB, the RBP1 subunit is ubiquitylated by NEDD4 [72] and the Elongin A ubiquitin ligase complex [73], inducing its extraction by VCP/p97 and subsequent proteasome degradation.
Role of Chromatin in NER Pathway
Activation of the NER pathway requires DNA not wrapped around histones, as various proteins need to access the double helix. UV damage to DNA provides the signal that leads to post-translational modifications of histones or ATP-dependent chromatin remodeling. These mechanisms allow the relaxation of chromatin around histones and increase the efficiency of the NER pathway [74,75].
Histone Modifications
Acetylation is mediated by histone acetyltransferases (HATs) and the reverse process, deacetylation, is catalyzed by deacetylases (HDACs). Histone acetylation promotes chromatin relaxation and activation, promoting DNA transcription [76]. Histone acetylation stimulates NER after UV damage [65]. Recent studies report that DDB2 interacts with HBO1 (HAT) in a UV-dependent manner leading to acetylation of H3 and H4, which increases chromatin accessibility [77]. DDB2 appears to be responsible for deacetylation of H3 and H4 through HDACs, leading to the stimulation of XPC recruitment [78]. Methylation acts differently on chromatin state depending on the number of methyl groups and the residues on which they are added [79]. Recent work has shown that DDB2 recruits the methyltransferase ASH1L to damaged DNA regions, leading to tri-methylation of histone H3K4. This process induces XPC binding to nucleosomes [80]. UV irradiation also stimulates tri-methylation of H3K79 by DOT1L, which also correlates with XPC recruitment. Deletion of DOT1L is present in many cases of melanoma [81]. Finally, another histone modification is phosphorylation which leads to a more relaxed state of chromatin and serves as a checkpoint in several processes, including DNA repair [82]. Phosphorylation of specific histones does not appear to affect the NER pathway; rather, it is the NER pathway that induces phosphorylation of histone H2AX via single-strand DNA production [83].
ATP-dependent Chromatin Remodeling
Chromatin remodelers are enzymes with an ATPase domain, which use ATP energy to modify the structure of nucleosomes. They are divided into 4 families: SWI/SNF, CHD, INO80, ISWI [84]. During NER, the SWI/SNF complex catalyzes the relaxation of chromatin, making it more accessible. CSB, which plays a key role in the NER pathway, belongs to this group [85]. An additional role of SWI/SNF is its association with XPC, which promotes the recruitment of subsequent repair factors [86]. The remodeler INO80 interacts at the level of damaged sites with DDB1, suggesting a role in XPC recruitment [87]. CHD is recruited following UV damage and mediates XPC binding to TFIIH [88].
Histone Chaperones
Proteins are involved in the transport and mobilization of histones at the chromatin level [89]. CAF-1 and HIRA are associated with NER, as they are recruited in the late stages of the pathway and are involved in the deposition of neo-synthesis histones after damage repair [90,91].
Conclusions
DDR pathways, including NER, play a key role in the formation of various tumor types and their sensitivity or resistance to treatment. The NER pathway is composed of a variety of processes that are finely regulated by each other and integrated with many other cellular pathways and alterations in this pathway play an important role in many tumor types, including STS. Recent studies contribute to the notion that NER pathway deficiencies constitute potential cancer therapeutic targets. It has been found that putative damaging germline and somatic alterations in NER genes were present in STS [14]. Moreover, recent findings provide novel insights into a synthetic lethal relationship between clinically observed NER gene deficiencies and sensitivity to irofulven and its potential synergistic combination with other drugs [9]. There is still little evidence on the association between cancer risk and variations in NER genes; in fact, there are no FDA-approved targeted therapies that target germline or somatic mutations in NER pathway genes. However, mutations in NER genes can have multiple roles as biomarkers: they can act as predictive biomarkers, indicating an increased risk of developing cancer, being useful for early cancer detection by subjecting the population to higher levels of screening; at the same time, they can also be used as prognostic biomarkers, giving precise indications to physicians about the degree of sensitivity to drugs targeting DNA repair deficiencies, if variations in NER genes are present. Recent studies focus instead on the therapeutic role of NER inhibitors, such as spironolactone [92] well as triptolide, which inhibits NER by affecting XPB and transcription. NER inhibition has been shown to reverse acquired resistance to alkylating agents in multiple myeloma cells [12]. Hence, it may be another adjunct target to be considered in combination therapies. Their further investigation in these tumor types is necessary for the identification of new biomarkers or therapeutic targets (Figures 1 and 2).
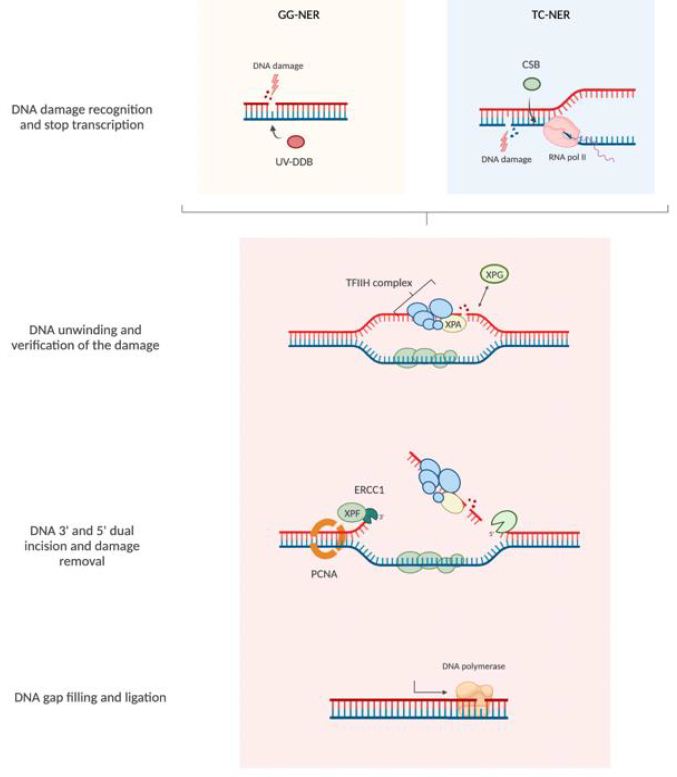
Figure 2: Nucleotide excision repair pathway (NER). NER pathways can be classified as either global genome repair (GGR) and transcription-coupled repair (TCR). The NER pathway consists of a series of reactions: recognition of DNA damage, unwinding double-stranded DNA in the neighborhood of the damage, excision of the damaged nucleotides, and filling the gap by DNA synthesis and ligation.
Author Contributions
Conceptualization, S.P.; writing—original draft preparation, F.S., A.P., A.B., E.C, S.F., O.C., E.G. and S.P; writing—review and editing, I.P, A.B. and G.S.; supervision, A.L. and S.P.; funding acquisition, I.P., D.A.C, S.P.; figures preparation: S.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Regione Toscana Bando Salute 2018, (Research project CUP n. D78D20000870002), grant number D78D20000870002.
Acknowledgments
Our special memory goes to Alessio Cerasola, an unforgettable guy who fights the disease with his ever-present smile.
Conflicts of Interest
The authors declare no conflict of interest
References
- Pillozzi et al. Soft Tissue Sarcoma: An Insight on Biomarkers at Molecular, Metabolic and Cellular Level. Cancers (Basel) [crossref]
- Gambale et al. Pharmacogenomics of soft tissue sarcomas: New horizons to understand efficacy and toxicity. Cancer Treat Res Commun. [crossref]
- H. Beck, R. B. West, e M. van de Rijn, Gene expression profiling for the investigation of soft tissue sarcoma pathogenesis and the identification of diagnostic, prognostic, and predictive biomarkers. Virchows Arch [crossref].
- Park, H. Kim, V. Hassebroek, Y. Azuma, C. Slawson, e M. Azuma, Chromosomal localization of Ewing sarcoma EWSR1/FLI1 protein promotes the induction of aneuploidy. J Biol Chem [crossref]
- Jiang et al. Alterations of DNA damage repair in cancer: from mechanisms to applications. Ann Transl Med .
- M. Scarbrough et al. A Cross-Cancer Genetic Association Analysis of the DNA repair and DNA Damage Signaling Pathways for Lung, Ovary, Prostate, Breast and Colorectal Cancer. Cancer Epidemiol Biomarkers Prev [crossref]
- Knijnenburg et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Reports [crossref]
- A. A. Castro, J. C. M. Mombach, R. M. C. de Almeida, e J. C. F. Moreira, Impaired expression of NER gene network in sporadic solid tumors. Nucleic Acids Res [crossref]
- Topka et al. Targeting Germline and Tumor Associated Nucleotide Excision Repair Defects in Cancer. Clin Cancer Res [crossref]
- Duan, J. Ulibarri, K. J. Liu, e P. Mao, Role of Nucleotide Excision Repair in Cisplatin Resistance. International Journal of Molecular Sciences [crossref]
- Tavecchio, C. Natoli, P. Ubezio, E. Erba, e M. D’Incalci, Dynamics of cell cycle phase perturbations by trabectedin (ET-743) in nucleotide excision repair (NER)-deficient and NER-proficient cells, unravelled by a novel mathematical simulation approach. Cell Prolif [crossref]
- Szalat et al. Nucleotide excision repair is a potential therapeutic target in multiple myeloma. Leukemia.
- Bialick et al. Pan-sarcoma analysis of DNA damage response pathway alterations and deficiency.. JCO, vol. 40, n. 16_suppl, pagg. 11548-11548, giu. 2022, doi: 10.1200/JCO.2022.40.16_suppl.11548.
- Pasqui et al. Alteration of the Nucleotide Excision Repair (NER) Pathway in Soft Tissue Sarcoma. Int J Mol Sci. [crossref]
- Spivak, Nucleotide excision repair in humans. DNA Repair (Amst), vol. 36, pagg. 13-18, dic. 2015, doi: 10.1016/j.dnarep.2015.09.003.
- Pietrasik, G. Zajac, J. Morawiec, M. Soszynski, M. Fila, e J. Blasiak, Interplay between BRCA1 and GADD45A and Its Potential for Nucleotide Excision Repair in Breast Cancer Pathogenesis. Int J Mol Sci [crossref]
- O. Black, Xeroderma Pigmentosum. Head Neck Pathol, vol. 10, n. 2, pagg. 139-144, giu. 2016, doi: 10.1007/s12105-016-0707-8.
- G. Sargent et al. Role of the nucleotide excision repair gene ERCC1 in formation of recombination-dependent rearrangements in mammalian cells. Nucleic Acids Res [crossref]
- -S. Rodrigo et al. Topoisomerase II-alpha protein expression and histological response following doxorubicin-based induction chemotherapy predict survival of locally advanced soft tissues sarcomas. Eur J Cancer [crossref]
- Italiano et al. ERCC5/XPG, ERCC1, and BRCA1 gene status and clinical benefit of trabectedin in patients with soft tissue sarcoma. Cancer [crossref]
- M. Kane et al. Correlation of High-Risk Soft Tissue Sarcoma Biomarker Expression Patterns with Outcome following Neoadjuvant Chemoradiation. Sarcoma [crossref]
- G. Clarkson e R. D. Wood, Polymorphisms in the human XPD (ERCC2) gene, DNA repair capacity and cancer susceptibility: an appraisal. DNA Repair (Amst). [crossref]
- H. Kraemer, J. J. DiGiovanna, e D. Tamura, Xeroderma Pigmentosum. in GeneReviews®, M. P. Adam, D. B. Everman, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. Bean, K. W. Gripp, e A. Amemiya, A c. di Seattle (WA): University of Washington, Seattle, 1993. Consultato: 1 settembre 2022. [Online]. Disponibile su: http://www.ncbi.nlm.nih.gov/books/NBK1397/
- S. Moura et al. A DNA damage repair gene-associated signature predicts responses of patients with advanced soft-tissue sarcoma to treatment with trabectedin. Mol Oncol [crossref]
- Franceschi et al. Whole-exome analysis of a Li-Fraumeni family trio with a novel TP53 PRD mutation and anticipation profile. Carcinogenesis [crossref]
- Manandhar, K. S. Boulware, e R. D. Wood, The ERCC1 and ERCC4 (XPF) genes and gene products. Gene. [crossref]
- H. Chan et al. Germline Mutations in Cancer Predisposition Genes are Frequent in Sporadic Sarcomas. Sci Rep [crossref]
- G. van Ravensteijn et al. Which angiosarcoma subtypes may benefit from immunotherapy?. JCO, vol. 40, n. 16_suppl, pagg. 11572-11572, giu. 2022, doi: 10.1200/JCO.2022.40.16_suppl.11572.
- Muniesa-Vargas, A. F. Theil, C. Ribeiro-Silva, W. Vermeulen, e H. Lans, XPG: a multitasking genome caretaker. Cell Mol Life Sci, vol. 79, n. 3, pag. 166, mar. 2022, doi: 10.1007/s00018-022-04194-5.
- S. Moura et al. CUL4A, ERCC5, and ERCC1 as Predictive Factors for Trabectedin Efficacy in Advanced Soft Tissue Sarcomas (STS): A Spanish Group for Sarcoma Research (GEIS) Study. Cancers (Basel) [crossref]
- Benna et al. Genetic susceptibility to bone and soft tissue sarcomas: a field synopsis and meta-analysis. Oncotarget. [crossref]
- van der Weegen et al. The cooperative action of CSB, CSA, and UVSSA target TFIIH to DNA damage-stalled RNA polymerase II. Nat Commun [crossref]
- Zhang et al. Mutations in UVSSA cause UV-sensitive syndrome and destabilize ERCC6 in transcription-coupled DNA repair. Nat Genet [crossref]
- Laugel, Cockayne syndrome: the expanding clinical and mutational spectrum. Mech Ageing Dev, vol. 134, n. 5-6, pagg. 161-170, giu. 2013, doi: 10.1016/j.mad.2013.02.006.
- T. Sherry, M. Ward, e K. Sirotkin, dbSNP—Database for Single Nucleotide Polymorphisms and Other Classes of Minor Genetic Variation. Genome Res., vol. 9, n. 8, pagg. 677-679, gen. 1999, doi: 10.1101/gr.9.8.677. [crossref]
- Groisman et al. CSA-dependent degradation of CSB by the ubiquitin-proteasome pathway establishes a link between complementation factors of the Cockayne syndrome. Genes Dev [crossref]
- Moslehi, H.-S. Tsao, N. Zeinomar, C. Stagnar, S. Fitzpatrick, e A. Dzutsev, Integrative genomic analysis implicates ERCC6 and its interaction with ERCC8 in susceptibility to breast cancer. Sci Rep [crossref]
- -J. Jing et al. Epistatic SNP interaction of ERCC6 with ERCC8 and their joint protein expression contribute to gastric cancer/atrophic gastritis risk. Oncotarget, vol. 8, n. 26, pagg. 43140-43152, giu. 2017, doi: 10.18632/oncotarget.17814.
- -F. Chiu et al. A novel single nucleotide polymorphism in ERCC6 gene is associated with oral cancer susceptibility in Taiwanese patients. Oral Oncol [crossref]
- Fadda, Role of the XPA protein in the NER pathway: A perspective on the function of structural disorder in macromolecular assembly. Comput Struct Biotechnol J [crossref]
- P. M. Melis, M. Luijten, L. H. F. Mullenders, e H. van Steeg, The role of XPC: implications in cancer and oxidative DNA damage. Mutat Res [crossref]
- J. Greber, D. B. Toso, J. Fang, e E. Nogales, The complete structure of the human TFIIH core complex. Elife.
- Grünberg, L. Warfield, e S. Hahn, Architecture of the RNA polymerase II preinitiation complex and mechanism of ATP-dependent promoter opening. Nat Struct Mol [crossref]
- Tirode, D. Busso, F. Coin, e J. M. Egly, Reconstitution of the transcription factor TFIIH: assignment of functions for the three enzymatic subunits, XPB, XPD, and cdk7. Mol Cell [crossref]
- Rimel e D. J. Taatjes, The essential and multifunctional TFIIH complex. Protein Sci. [crossref]
- Coin, V. Oksenych, V. Mocquet, S. Groh, C. Blattner, e J. M. Egly, Nucleotide excision repair driven by the dissociation of CAK from TFIIH. Mol Cell. [crossref]
- Lehmann, DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie .
- Hu et al. Polymorphisms in the two helicases ERCC2/XPD and ERCC3/XPB of the transcription factor IIH complex and risk of lung cancer: a case-control analysis in a Chinese population. Cancer Epidemiol Biomarkers Prev [crossref]
- Ma, Y. Zhang, T. S. Sun, e J. H. Yao, Role of ERCC2 and ERCC3 gene polymorphisms in the development of osteosarcoma. Genet Mol Res, vol. 15, n. 1, mar. 2016, doi: 10.4238/gmr.15017302.
- Zhang, S.-Y. Gu, P. Zhang, Z. Jia, e J.-H. Chang, ERCC2 Lys751Gln polymorphism is associated with lung cancer among Caucasians. Eur J Cancer, vol. 46, n. 13, pagg. 2479-2484, set. 2010, doi: 10.1016/j.ejca.2010.05.008.
- Ni et al. Association of ERCC1 and ERCC2 polymorphisms with colorectal cancer risk in a Chinese population. Sci Rep, vol. 4, pag. 4112, feb. 2014, doi: 10.1038/srep04112.
- Le Morvan et al. Genetic polymorphisms of the XPG and XPD nucleotide excision repair genes in sarcoma patients. Int J Cancer. [crossref]
- Varshavsky, The ubiquitin system. Trends in Biochemical Sciences, vol. 22, n. 10, pagg. 383-387, ott. 1997, doi: 10.1016/S0968-0004(97)01122-5.
- Zheng e N. Shabek, Ubiquitin Ligases: Structure, Function, and Regulation. Annu Rev Biochem, vol. 86, pagg. 129-157, giu. 2017, doi: 10.1146/annurev-biochem-060815-014922.
- P. Jackson e D. Durocher, Regulation of DNA damage responses by ubiquitin and SUMO. Mol Cell, vol. 49, n. 5, pagg. 795-807, mar. 2013, doi: 10.1016/j.molcel.2013.01.017.
- Kulathu e D. Komander, Atypical ubiquitylation – the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat Rev Mol Cell Biol [crossref]
- E. Reyes-Turcu, K. H. Ventii, e K. D. Wilkinson, Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem, vol. 78, pagg. 363-397, 2009, doi: 10.1146/annurev.biochem.78.082307.091526.
- Bergink e S. Jentsch, Principles of ubiquitin and SUMO modifications in DNA repair. Nature [crossref]
- Groisman et al. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell [crossref]
- K. Chauhan, Y. Sun, Q. Zhu, e A. A. Wani, Timely upstream events regulating nucleotide excision repair by ubiquitin-proteasome system: ubiquitin guides the way. DNA Repair (Amst) [crossref]
- Chitale e H. Richly, Timing of DNA lesion recognition: Ubiquitin signaling in the NER pathway. Cell Cycle [crossref]
- Zhang e F. Gong, The emerging role of deubiquitination in nucleotide excision repair. DNA Repair (Amst), vol. 44, pagg. 118-122, ago. 2016, doi: 10.1016/j.dnarep.2016.05.035.
- Matsumoto et al. Functional regulation of the DNA damage-recognition factor DDB2 by ubiquitination and interaction with xeroderma pigmentosum group C protein. Nucleic Acids Res [crossref]
- Robu, R. G. Shah, N. Petitclerc, J. Brind’Amour, F. Kandan-Kulangara, e G. M. Shah, Role of poly(ADP-ribose) polymerase-1 in the removal of UV-induced DNA lesions by nucleotide excision repair. Proc Natl Acad Sci U S A [crossref]
- -A. Lee et al. BAP1 promotes the repair of UV-induced DNA damage via PARP1-mediated recruitment to damage sites and control of activity and stability. Cell Death Differ [crossref]
- Ribeiro-Silva et al. Ubiquitin and TFIIH-stimulated DDB2 dissociation drives DNA damage handover in nucleotide excision repair. Nat Commun [crossref]
- Sugasawa et al. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell. [crossref]
- van Cuijk et al. SUMO and ubiquitin-dependent XPC exchange drives nucleotide excision repair. Nat Commun [crossref]
- Wei et al. BRCA1 contributes to transcription-coupled repair of DNA damage through polyubiquitination and degradation of Cockayne syndrome B protein. Cancer Sci [crossref]
- Nicholson e K. G. Suresh Kumar, The multifaceted roles of USP7: new therapeutic opportunities. Cell Biochem Biophys [crossref]
- Sarasin, UVSSA and USP7: new players regulating transcription-coupled nucleotide excision repair in human cells. Genome Med [crossref]
- Anindya, O. Aygün, e J. Q. Svejstrup, Damage-induced ubiquitylation of human RNA polymerase II by the ubiquitin ligase Nedd4, but not Cockayne syndrome proteins or BRCA1. Mol Cell, vol. 28, n. 3, pagg. 386-397, nov. 2007, doi: 10.1016/j.molcel.2007.10.008.
- Yasukawa, T. Kamura, S. Kitajima, R. C. Conaway, J. W. Conaway, e T. Aso, Mammalian Elongin A complex mediates DNA-damage-induced ubiquitylation and degradation of Rpb1. EMBO J [crossref]
- -R. Duan e M. J. Smerdon, UV damage in DNA promotes nucleosome unwrapping. J Biol Chem [crossref]
- Mohan, C. Das, e J. Tyler, Histone and Chromatin Dynamics Facilitating DNA repair. DNA Repair (Amst), vol. 107, pag. 103183, nov. 2021, doi: 10.1016/j.dnarep.2021.103183.
- C. Hodawadekar e R. Marmorstein, Chemistry of acetyl transfer by histone modifying enzymes: structure, mechanism and implications for effector design. Oncogene [crossref]
- Niida et al. Phosphorylated HBO1 at UV irradiated sites is essential for nucleotide excision repair. Nat Commun [crossref]
- Apelt, H. Lans, O. D. Schärer, e M. S. Luijsterburg, Nucleotide excision repair leaves a mark on chromatin: DNA damage detection in nucleosomes. Cell Mol Life Sci [crossref]
- L. Greer e Y. Shi, Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet [crossref]
- Balbo Pogliano, M. Gatti, P. Rüthemann, Z. Garajovà, L. Penengo, e H. Naegeli, ASH1L histone methyltransferase regulates the handoff between damage recognition factors in global-genome nucleotide excision repair. Nat Commun [crossref]
- Zhu et al. The protective role of DOT1L in UV-induced melanomagenesis. Nat Commun [crossref]
- Rossetto, N. Avvakumov, e J. Côté, Histone phosphorylation: a chromatin modification involved in diverse nuclear events. Epigenetics. [crossref]
- Matsumoto et al. Perturbed gap-filling synthesis in nucleotide excision repair causes histone H2AX phosphorylation in human quiescent cells. J Cell Sci [crossref]
- G. Wang, C. D. Allis, e P. Chi, Chromatin remodeling and cancer, Part II: ATP-dependent chromatin remodeling. Trends Mol Med [crossref]
- Citterio et al. ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol Cell Biol [crossref]
- Zhao et al. Modulation of nucleotide excision repair by mammalian SWI/SNF chromatin-remodeling complex. J Biol Chem, vol. 284, n. 44, pagg. 30424-30432, ott. 2009, doi: 10.1074/jbc.M109.044982.
- Jiang et al. INO80 chromatin remodeling complex promotes the removal of UV lesions by the nucleotide excision repair pathway. Proc Natl Acad Sci U S A, vol. 107, n. 40, pagg. 17274-17279, ott. 2010, doi: 10.1073/pnas.1008388107.
- Rüthemann, C. Balbo Pogliano, T. Codilupi, Z. Garajovà, e H. Naegeli, Chromatin remodeler CHD1 promotes XPC-to-TFIIH handover of nucleosomal UV lesions in nucleotide excision repair. EMBO J [crossref]
- Avvakumov, A. Nourani, e J. Côté, Histone chaperones: modulators of chromatin marks. Mol Cell [crossref]
- Adam e S. E. Polo, Chromatin dynamics during nucleotide excision repair: histones on the move. Int J Mol Sci [crossref]
- Bouvier, J. Ferrand, O. Chevallier, M. T. Paulsen, M. Ljungman, e S. E. Polo, Dissecting regulatory pathways for transcription recovery following DNA damage reveals a non-canonical function of the histone chaperone HIRA. Nat Commun [crossref]
- Alekseev, M. Ayadi, L. Brino, J.-M. Egly, A. K. Larsen, e F. Coin, A small molecule screen identifies an inhibitor of DNA repair inducing the degradation of TFIIH and the chemosensitization of tumor cells to platinum. Chem Biol [crossref]