Abstract
The brains from two patients that died with Alzheimer’s disease were examined to determine the role of mTOR and ubiquitin in the pathogenesis of beta-amyloid plaque and Tau-tangle formation. The brain cortex was examined H&E sections, Sevier-Munger silver staining, and IHC stains for B-amyloid, tau, and ubiquitin. Beta-amyloid plaques and Tau-tangles were then double stained for mTOR and ubiquitin to identify their association of Alzheimer’s disease in clinical specimens. In beta-amyloid plaques and Tau-tangles, and in neurons associated with beta-amyloid plaque and Tau-tangles, we observed aggregation of both mTor and ubiquitin. The presence of ubiquitin in aggresomes has been observed in neurodegenerative diseases previously and correlates with proteasome dysfunction. Likewise, increased mTOR signaling has been associated with decreased autophagy and has been observed in Alzheimer’s models. Both of these important molecules were found to co-localize in the aggresomes in lesional areas in the Alzheimer’s brains in our study.
Keywords
Alzheimer’s disease, mTOR, proteasome, aggresome, beta-amyloid
Introduction
Protein synthesis and protein degradation are under tight control in most cells of the body. Under normal homeostatic conditions, misfolded proteins and damaged cellular organelles are cleared by either the unfolded protein response (UPR) or autophagy [1]. The unfolded protein response can down-regulate overall protein synthesis while up-regulating molecular chaperons and stress response genes. Soluble unfolded proteins are targeted to the juxta-nuclear quality control (JUNQ) where folding chaperones can promote refolding or the proteins are degraded by the proteasome [2]. On the other hand, insoluble proteins are targeted to the lysosome via autophagy, and if this fails aggresomes form [2].
The liver has one of the highest protein production and turnover rates of any organ in the body given that it must produce all of the albumin formed as well as the clotting factors found in plasma [1]. It is well known in liver pathology that impairment of metabolism can lead to failure of the protein quality control mechanism, misfolding of proteins, and accumulation of intra-cellular protein aggregates termed Mallory-Denk bodies (MDBs) such as are seen in non-alcoholic steatohepatitis (NASH), Hepatitis C infection, hepatocellular carcinoma, primary biliary cirrhosis, Wilson’s disease, a beta-lipoproteinemia, and alcoholic liver disease (ALD) [2]. Conditions of ongoing stress can lead to a failure of normal homeostatic mechanisms, mTOR signaling dysregulation, and accumulation of protein aggregates including cytokeratins covalently bound to multimers of ubiquitin [2, 3]. Ubiquitin is a signal protein used to target proteins to the proteasome, however these Cytokeratin-ubiquitin aggregates lead to ubiquitin-proteasome dysfunction and accumulation of protein aggregates [2, 3].
By contrast, neurons do not have the same synthetic demands as liver cells, however they are highly metabolically active cells which must, in some cases, transport signals over long distances [1, 4]. Neurons also do not undergo mitosis and are rarely replaced [1]. These limitations make the process of autophagy similarly important for neurons survival [1, 5]. Failure of protein clearance, likely through autophagy, can lead to protein aggregation as seen in neurodegenerative conditions such as Alzheimer’s disease and Parkinson’s disease [5, 6]. The early pathogenesis of Alzheimer’s disease correlates with ubiquitin-proteasome failure [6-8]. Later in the pathogenesis of Alzheimer’s disease, ubiquitin bound to proteins accumulates in aggregates [6-8]. Finally, ubiquitin is known to accumulate in beta-amyloid plaques and Tau-tangles co-localize with ubiquitin which is covalently bound to proteins in the insoluble complexes [9].
There is abundant support in the literature that indicates that mTOR plays a role in beta-amyloid plaque and Tau-tangle formation in Alzheimer’s disease as well [10]. The mTOR signaling complexes mTorc1 and mTorc2 respond to cellular changes in homeostasis including nutritional status and growth factor signaling [4, 10, 11]. Increased mTOR signaling leads to a decrease in the autophagosome mediated degradation of proteins [10-13]. Early in the pathogenesis of Alzheimer’s disease soluble misfolded proteins are targeted to the UPR for refolding, but as protein accumulates, the proteins are bound with ubiquitin and targeted for autophagy [12-14]. Increased levels of mTOR signaling block the autophagy process and this leads to further accumulation of proteins, including mTOR, and these proteins form the large aggregates characteristic of Alzheimer’s disease [14-17].
Methods
Formalin-fixed, Paraffin-embeded tissue blocks and slides were obtained from autopsies obtained at the Harbor-UCLA hospital. The neuropathologic evaluation at autopsy evaluated midbrain, medulla, pons, cerebellum and denate nucleus, thalamus, basal ganglia, cingulate gyrus, frontal cortex, temporal cortex, and occipital lobe with H&E staining. Sections were also evaluated with modified Bielschowsky silver staining. Standard H&E, Sevier-Munger, and IHC stained slides of the hippocampus, temporal, and frontal lobes were reviewed along with the original neuropathology reports. Standard HRP-IHC was performed for antibodies to ubiquitin, amyloid, and tau. Additionally, IHC immunofluorescence for mTOR and ubiquitin was performed. The slides were double stained with commercially obtained antibodies to mTOR and ubiquitin, and visualized on a fluorescent microscope.
Observations
For our study we utilized autopsy brain tissue from two clinically and pathologically confirmed cases of Alzheimer’s disease. Review of records showed a neuropathologic reports identified cases which are ranked per NIA-AA along three parameters amyloid plaque, neurofibrillary tangle, and neuritic plaque. Although both cases had clinical records indicating cognitive impairment without other explanation, neuropathologic assessment was scored as A3, B3, C3 or “High” level of neuropathologic change, which is considered sufficient for an explanation of dementia [18]. Alzheimer’s patients showed co-localization of mTOR and ubiquitin within cells associate with both beta-amyloid plaques and Tau-tangles. To our knowledge, this is the first time in which mTor and has been shown to co-localize with ubiquitin in beta-amyloid plaques and neurofibrillary Tau-tangles.
We identified beta-amyloid plaques and Tau-tangles in two patients in the hippocampus consistent with previous neuropathologic diagnoses of Alzheimer’s dementia. These patients for example, in patient 1 (Figure 1, A) shows a beta-amyloid plaque with IHC stain for B-amyloid. In Figure 1 (C), Sevier-Munger silver stain highlights Tau-tangles, including several Tau-tangles which are noted to run along a prominent axon. In figure 1 (B), immunofluorescence highlights mTOR (green) in a perinuclear distribution and and ubiquitin (red) in extra cellular clusters. In Figure 1 (D), several neurons associated with Tau-tangles were noted to show colocalization of mTOR and ubiquiting within cells. These cells showed co-localization of mTOR and cytoplasmic-ubiquitin and extensive cytoplasmic perinuclear ubiquitin with co-localized mTOR.
Similarly, in patient two (Figure 2) we observed beta-amyloid plaques stained with IHC to B-amyloid (A) and aggregates of protein within beta-amyloid plaques contain mTOR and ubiquitin (B). Part C shows neurofibrillary Tau-tangles with Sevier-Munger stain. Part D shows two cells which have colocalization of mTOR and ubiquitin. In these cells the ubiquitin stains red and was found within the cytoplasm, while the mTOR (green) is polarized to one side of the cell, and is seen primarily as yellow due to co-localization of mTOR and ubiquitin. These proteins accumulated within in the diseased neurons associated with beta-amyloid plaques and Tau-tangles, and can be found in the beta-amyloid plaques and Tau-tangles of later stages.
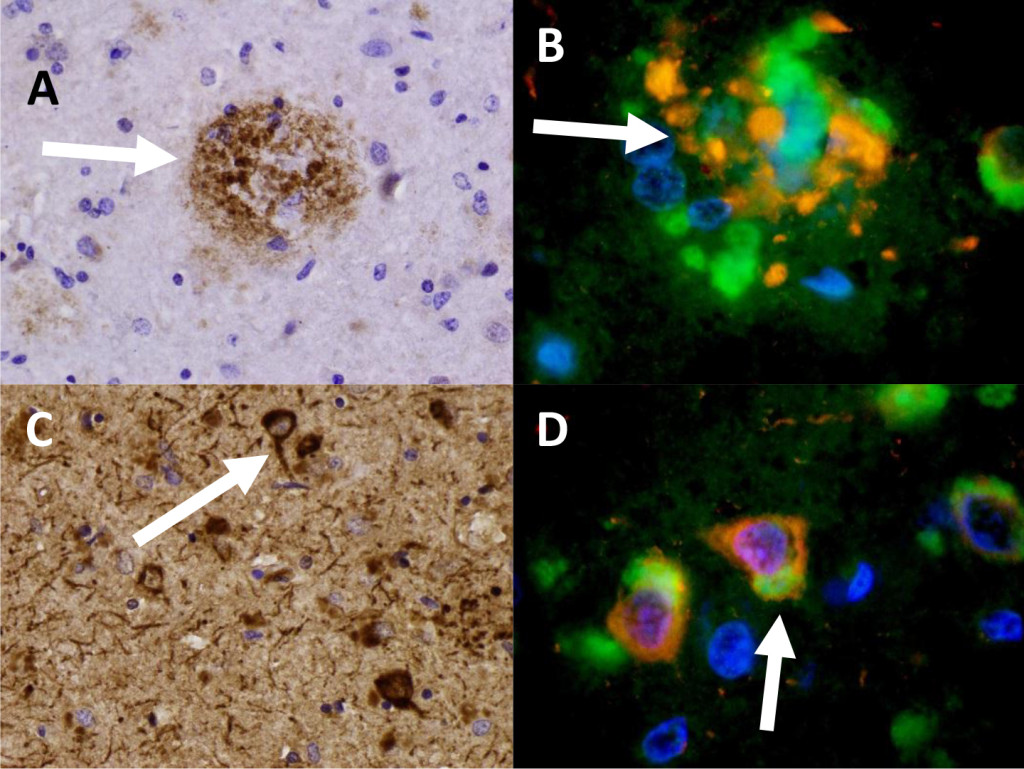
Figure 1. Patient 1 (A) Beta-amyloid plaque with IHC staining of B-amyloid. (B) A beta-amyloid plaque in Patient 1 specimen triple-stained for DAPI (blue) , Ubiquitin (red), and mTOR (green). 959x (C) Neurofibrillary Tau-tangles with IHC staining for Tau. 436x (D) Nerve cells (Patient 1) showing aggregation of ubiquitin (red) and mTOR (green). 950x
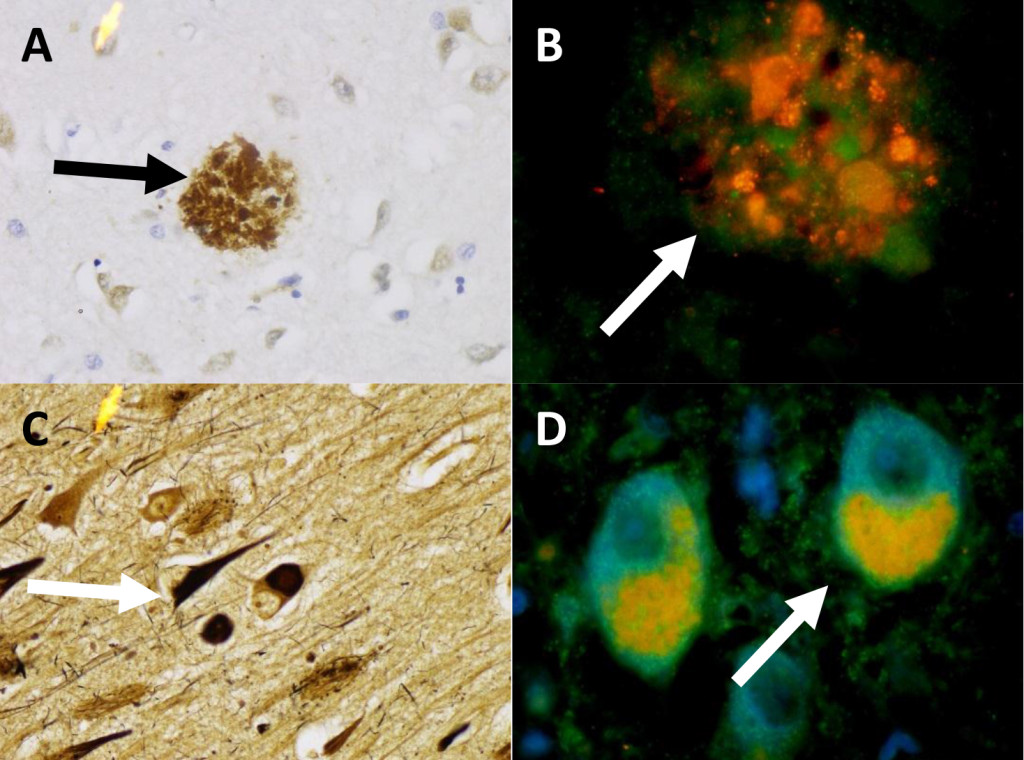
Figure 2. Patient 2 (A) Beta-amyloid plaque with IHC stain for Beta-amyloid. 436x (B) A Beta-amyloid plaque in Patient 2 double stained for Ubiquitin (red), and mTOR (green). 959x (C) Patient 2 Sevier-Munger stain highlighting Tau-tangles. 436x (D) Nerve cells with colocalization of mTOR (green) and ubiquitin (red) in neurons with Tau-tangle formation. 959x
Conclusions
For the first time, we have demonstrated that mTOR and ubiquitin co-localize to beta-amyloid plaques and Tau-tangles found in Alzheimer’s disease. These findings further supports the already existing evidence that the mTOR/autophagy system is likely involved in the pathogenesis of Alzheimer’s disease, and perhaps is involved in other neurodegenerative diseases as well. Protein homeostasis including synthesis, folding, and degradation are essential to their cellular function and must be maintained under tight control. Dysfunction at any of the steps in these pathways can lead to a backup of the cyclic cellular assembly line and the development of additional dysfunction that is not easily resolved. It will be interesting to determine the way each of these pathways fit together in vivo for a wide range of neurologic conditions and how these processes are similar and dissimilar between organ systems. Our future efforts will be directed at further characterization of the molecular pathogenesis of protein misfolding and mTor signaling in patients with Alzheimer’s disease.
References
- Kumar V, Abbas AK, Aster JC (2015) Robbins and Cotran pathologic basis of disease (9th edn). Philadelphia, Elsevier/Saunders, USA.
- Afifiyan N, Tillman B, French BA, Masouminia M, Samadzadeh S, et al. (2017) Over expression of proteins that alter the intracellular signaling pathways in the cytoplasm of the liver cells forming Mallory-Denk bodies. Exp Mol Pathol 102: 106–114. [crossref]
- Hay N, Sonenberg N (2004) Upstream and downstream of mTOR. Genes Dev 18: 1926–1945. [crossref]
- French SW, Mendoza AS, Peng Y(2016) The mechanisms of Mallory-Denk body formation are similar to the formation of aggresomes in Alzheimer’s disease and other neurodegenerative disorders. Exp Mol Pathol 100: 426–433. [crossref]
- Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR5, et al. (2014) Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol112: 24–49. [crossref]
- Perluigi M, Domenico DF, Butterfield DA (2015) mTOR signaling in aging and neurodegeneration: At the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol Dis 84: 39–49. [crossref]
- Li Q, Liu Y, Sun M (2017) Autophagy and Alzheimer’s Disease. Cell Mol Neurobiol 37: 377–388. [crossref]
- Zare-Shahabadi A, Masliah E, Johnson GV, Rezaei N (2015) Autophagy in Alzheimer’s disease. Rev Neurosci 26: 385–395. [crossref]
- Love S (2015) Greenfields neuropathology. Boca Raton: CRC Press, USA.
- Cai Z, Chen G, He W, Xiao M, Yan LJ (2015) Activation of mTOR: a culprit of Alzheimer’s disease? Neuropsychiatr Dis Treat 11: 1015–1030. [crossref]
- Chong ZZ, Shang YC, Wang S, Maiese K (2012) A Critical Kinase Cascade in Neurological Disorders: PI 3-K, Akt, and mTOR. Future Neurol 7: 733–748. [crossref]
- Pei JJ, Hugon J (2008) mTOR-dependent signalling in Alzheimer’s disease. J Cell Mol Med 12: 2525–2532. [crossref]
- O’ Neill C (2013) PI3-kinase/Akt/mTOR signaling: impaired on/off switches in aging, cognitive decline and Alzheimer’s disease. Exp Gerontol 48: 647–653. [crossref]
- Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Eikelenboom P, et al. (2009) The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am J Pathol 174, 1241–1251. [crossref]
- Switon K, Kotulska K, Janusz-Kaminska A, Zmorzynska J, Jaworski J (2017) Molecular neurobiology of mTOR. Neuroscience 341: 112–153. [crossref]
- Talboom JS, Velazquez R, Oddo S (2015) The mammalian target of rapamycin at the crossroad between cognitive aging and Alzheimer’s disease. NPJ Aging Mech Dis 1: 15008. [crossref]
- Yates SC, Zafar A, Hubbard P, Nagy S, Durant S, et al. (2013) Dysfunction of the mTOR pathway is a risk factor for Alzheimer’s disease. Acta Neuropathol Commun 1: 3. [crossref]
- Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, et al. (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8: 1–13. [crossref]