Abstract
The finding that autoantibodies against glutamic acid decarboxylase that synthesizes GABA, provoke Diabetes type I, draws attention on a GABAergic regulation of the endocrine pancreas suppressing catabolic glucagon release if anabolic insulin is liberated; a GABA deficiency would then impair this exclusion mechanism, allowing a release of both hormones. Moreover, the GABA deficiency alters a mechanism terminating insulin release; an insulin leakage renders differentiated cells gradually resistant to insulin, while responding to glucagon. Mitotic cells with new insulin receptors, respond to both hormones, displaying a hybrid metabolic pattern typically found in tumor cells. They cannot get their mitochondrial acetyl CoA from glycolysis, since pyruvate kinase and pyruvate dehydrogenase are OFF, following the glucagon signal. Nor can they form acetyl CoA by the beta-oxidation of fatty acids, since the insulin signal they receive elicits the synthesis of fatty acids, which automatically closes their beta-oxidation. Indeed, malonyl CoA produced along the lipogenic pathway inhibits the mitochondrial carnityl-transporter of fatty acids. Hence, with both the glycolytic and fatty acid sources of mitochondrial acetyl CoA closed, tumor cells can only get their vital mitochondrial acetyl CoA supply from the ketolysis of ketone bodies.
Differentiated tissues resistant to insulin, but responding to glucagon, adequately provide these ketone bodies. The enzyme, Succinyl-CoA: 3-oxoacid-CoA transferase (SCOT) is specific to ketolysis producing acetyl CoA. Its inhibition deprives tumor cell mitochondria of acetyl CoA, which should hold back tumor development. Inhibiting also the cytosolic acetyl CoA synthetase that tumor cells use for feeding their lipid synthesis should block the tumor.
Keywords
Endocrine pancreas, tumor cell metabolism, Ketone bodies dependency, SCOT inhibition, Acetyl CoA synthetase inhibition
Introduction
Insulin dependent Diabetes (type 1) may result from an attack of pancreatic beta cells, by autoantibodies against, the enzyme synthetizing GABA [1]. We do know that pancreatic beta cell co-release insulin and GABA, insulin release corrects hyperglycemia, while GABA will inhibit, via GABA A ionotropic receptors, neighboring delta and alpha cells, respectively releasing somatostatin and glucagon. Hence, the release of anabolic insulin is associated to a mechanism switching off, via GABA, catabolic glucagon release and somatostatin release [2, 3]. Moreover, GABA acts on beta cell auto-receptors (metabotropic GABA B) for putting an end to insulin release [4]. A GABA deficiency will then fail to completely, turn–off insulin release, the resulting leakage of insulin will desensitize in the long-run insulin receptors in differentiated cells (an effect reminding Type II diabetes or metabolic syndrome) [5]. The deficiency of GABA release also affects the mutual exclusion process of catabolism when anabolism takes place, sending a dual message to cells. Differentiated tissues resistant to insulin will take the catabolic part of this message and develop a neoglucogenic and ketogenic metabolism. Whereas cells that are not resistant to insulin, will receive both insulin and glucagon messages; these are new mitotic stem cells, with new insulin receptors, not yet affected by the chronic desensitization provoked by insulin leakage. These cells will rewire their metabolism as one observes for tumor cells [6-10].
Metabolic rewiring in cancer: A consequence of mixed pancreatic signals
Tumor cells are avid for glucose, and insulin elicits the incorporation of glucose transporters in their membrane. Indeed, following the binding of insulin to its tyrosine kinase receptor and the activation of MAP and PI3 kinases a downstream effect stimulates a phospholipase forming inositol 3, 4, 5 Phosphate (IP3) and diacyl glycerol (DAG). Then, an IP3-mediated calcium release from the reticulum in the cytosol triggers the exocytotic incorporation of glucose transporters in the cell membrane. In parallel, glycolysis increases because the decline of cAMP, elicited by a calcium-activated phosphodiesterase, cancels the inhibitory action of cAMP over fructose 2, 6 bis P synthesis, which increases this activator of glycolysis; incidentally, drugs like rolipram that inhibits the phosphodiesterase, would keep up cAMP and limit the glycolytic flux. In spite of the avid glucose influx, the phosphorylation of pyruvate kinase (PK) and pyruvate dehydrogenase (PDH), which inhibits these enzymes, closes the last steps of this increased glycolysis of tumor cells. In fact, tumor cells express the M2 form of PK that gives inactive dimers rather than active tetramers, forming a “bottleneck” at the end of the glycolytic pathway [11, 12]; which switches off the glycolytic production of acetyl CoA by PDH. The PK bottleneck increases the influx of substrates into the pentose pathway, while phosphoenolpyruvate (PEP) accumulated above the PK bottleneck, will rather convert to oxaloacetate (OAA) via PEP carboxykinase (PEPCK). What maintains this phosphorylation of PK and PDH? Presumably, the inhibitor CPI 17 of their phosphatase is synthetized via a stimulation of protein kinase C (PKC) by DAG [13]. We indeed know that PKC stimulates the production of this inhibitor, which cancels the calcium-dependent activation of the phosphatase PP1 by calcineurin that normally inactivates another inhibitor I1, of PP1 phosphatase. Then what keeps DAG elevated? We know that growth hormone stimulates adipose triglyceride lipase (ATGL), which produces an excess of DAG. The stimulation of growth hormone is itself a consequence of the GABA deficiency, because it is not limited to the pancreas. Low GABA increases epinephrine release from the adrenals, which inhibits somatostatin release. Consequently, growth hormone and IGF increase. In this respect, ATGL inhibitors would deserve a try [14]. Inhibition of growth hormone, DAG and PKC should also be interesting (remember that DAG acts on PKC similarly to phorbol ester carcinogens).
In sum, the glycolytic source of acetyl CoA at the entry of the Krebs cycle is well closed. On the other hand, the insulin anabolic action boosts synthetic processes and fatty acid synthesis, needed for making new membranes for mitotic cells. In this pathway malonyl CoA, the product of acetyl CoA carboxylase (ACC) at the beginning of the pathway, inhibits the transport and degradation of fatty acids in mitochondria, closing the beta-oxidation pathway, and the fatty acid source of acetyl CoA. Indeed, when fatty acid synthesis operates, the degradation of fatty acids automatically stops. Hence, if the two sources of mitochondrial acetyl CoA (glycolytic or fatty acid) are closed, mitotic stem cells will have to get their acetyl CoA from the ketolysis of ketone bodies, coming from the liver or other tissues selectively responding to catabolic hormones. Thus, with acetyl CoA that comes from ketolysis and (OAA) coming from phophoenol pyruvate (PEP), via PEP carboxykinase (PEPCK) and other sources, the citrate condensation takes place, starting the Krebs cycle [15, 16].
Ketogenesis and tumor cell ketolysis
We represent in Figure 1, a hepatocyte (left), responding to glucagon, it produces glucose and ketone bodies; the beta-oxidation of fatty acids is active, they enter in the mitochondria via the fatty acid acyl carnitine transporter, and will produce intra mitochondrial acetyl CoA. Then, four enzymes indicated in the figure 1, support ketogenesis, they lead to beta hydroxybutyrate, after the reduction of acetoacetate; the latter also degrades into acetone and CO2 (not represented). In catabolism, ketogenic amino acids such as leucine and a few others, also feed the ketogenic pathway with acetyl CoA and acetoacetyl CoA, leading to acetoacetate and beta hydroxybutyrate. The released beta hydroxy butyrate enters the tumor cell (right), through a transporter. It then reaches the mitochondria, where three enzymes indicated in the figure 1, will support ketolysis, resulting in acetyl CoA; the enzyme succinyl CoA 3- oxoacid- CoA transferase (SCOT) is the specific step. This ketolytic source of acetyl CoA is vital for tumor cells, since the other sources of mitochondrial acetyl CoA: the glycolytic (via PK and PDH), or the beta-oxidation of fatty acids, are both not working in tumor cells as described above. Thus, only ketone bodies provide acetyl CoA (via SCOT) to the citrate condensation reaction that starts the Krebs cycle. The citrate condensation is particularly active; but the citrate will quit the mitochondria and enter in the lipogenic pathway in the cytosol, forming fatty acids and lipids that are essential to build new membranes for mitotic cells. Thus blocking SCOT, the citrate efflux; and ATP citrate lyase, should hinder tumor development.
Tumor cells can still save their situation because they have in the cytosol an enzyme, acetyl CoA synthetase directly converting incorporated acetate into acetyl CoA [17]. Incidentally, we know that cholinergic neuromuscular synapses use acetate in preference to pyruvate for making acetyl CoA via this same enzyme, and then the acetyl moiety of acetylcholine, whereas brain cholinergic synapses use pyruvate rather than acetate for synthesizing the acetyl moiety of acetylcholine [18]. In tumor cells, acetyl CoA synthetase converts incorporated acetate into acetyl CoA, which enters in the fatty acids synthesis and the lipogenic pathways; in parallel, acetyl CoA forms acetoacetyl CoA in the cytosol via a cytosolic thiolase, and feeds the cholesterol-synthesizing pathway. In addition, we also find an acetoacetyl CoA synthetase activity in tumor cells. The acetyl and acetoacetyl CoA synthetases are enzymes forming adenylate intermediates before acetylation or acylation; inhibitors for these adenylating enzymes are available.
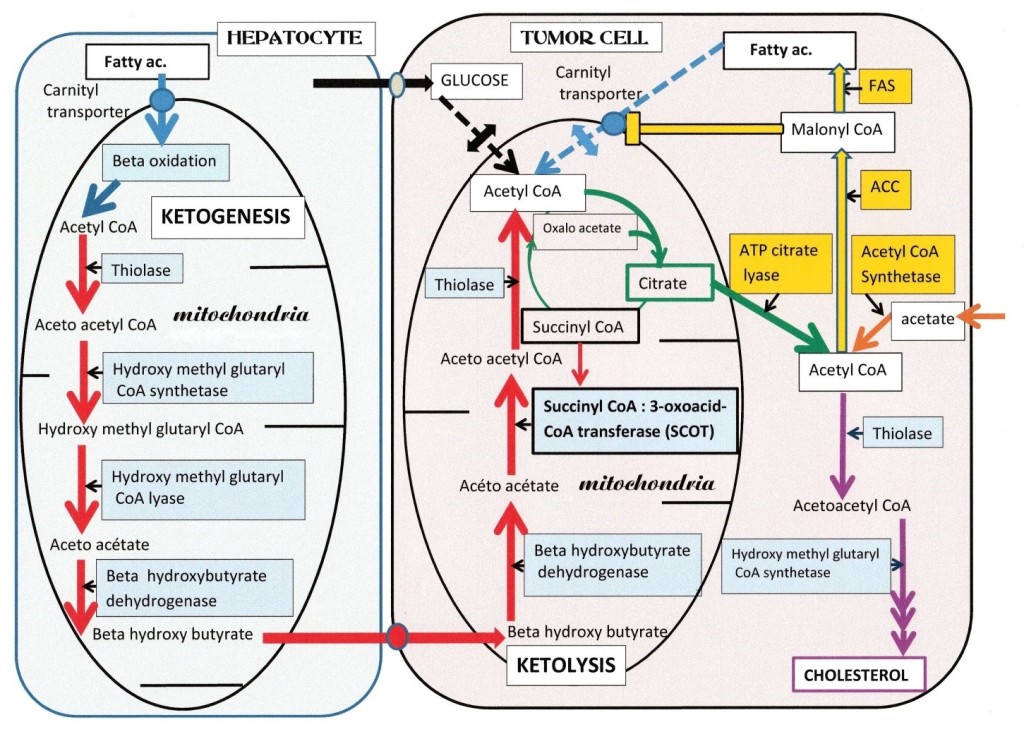
Figure 1.
Figure 1: Ketone bodies are vital for tumor cells. On the right, a tumor cell has a single ketolytic supply of mitochondrial acetyl CoA, of the three enzymes that form the pathway, Succinyl-CoA: 3-oxoacid-CoA transferase (SCOT) is specific for ketolysis. Note that the glycolytic and fatty acid sources of acetyl CoA are both interrupted; on the glycolytic side, pyruvate kinase and pyruvate dehydrogenase are OFF in tumors; and on the fatty acid side, their degradation will stop since fatty acid synthesis is active; indeed, the malonyl CoA intermediate, closes the carnityl transporter of fatty acids and beta oxidation. The citrate efflux in the cytosol feeds ATP citrate lyase, giving back acetyl CoA and oxaloacetate. Acetyl CoA follows the synthesis pathway of fatty acids, via acetyl CoA carboxylase (ACC) and fatty acid synthetase (FAS). The ACC forms the malonate inhibitor of fatty acid degradation. Tumor cells possess an acetyl CoA synthetase that converts the exogenous acetate influx into acetyl CoA, supporting the lipogenic pathway and the synthesis of acetoacety CoA via a cytosolic thiolase. Tumor cells also have an acetoacetyl CoA synthetase feeding the cholesterol synthesis pathway. On the left, a differentiated cell, a hepatocyte, resistant to insulin but responding to catabolic glucagon; produces glucose and ketone bodies. Fatty acids enter in the mitochondria via their carnityl transporter, and give by beta-oxidation acetyl CoA. The latter follows the ketogenic pathway, which consists of four enzymes, indicated in the figure. Ketogenesis forms acetoacetate and beta hydroxybutyrate. After release, beta hydroxybutyrate enters the tumor cell via a transporter; then in the tumor cell mitochondria, to give acetyl CoA by ketolysis; SCOT is the specific step, supporting this vital supply of acetyl CoA to the tumor cell, is its vulnerable point.
Color-code, in hepatocyte Beta-oxidation (blue arrow) ketogenesis (red arrows). In tumor cell: Interrupted Beta-oxidation and glycolytic supplies of acetyl CoA (dotted blue and black arrows) interrupted by small double arrows. Red arrows show ketolytic steps, (same enzymes as ketogenesis) except SCOT. Acetyl CoA starts the Krebs cycle, citrate release, ATP citrate lyase (green arrows). Salvage pathway: acetyl CoA synthetase (orange arrow), Lipogenic route (yellow arrows) and Cholesterol synthesis indicated by mauve arrows.
The ketogenic diet might not be a good idea
Initially, the ketogenic diet aimed to decrease the supply of glucose to tumors by replacing the glucose by ketone bodies, our other nutrient, provided by a high fat diet. However, in spite of several encouraging clinical reports, it seems that the ketogenic diet may not be a good idea, since ketone bodies are the only way for tumor cells to get their mitochondrial acetyl CoA. Several other works indicate that the utilization of ketone bodies drives tumor growth and metastasis; [19, 20] this agrees with the present work indicating that tumor cell metabolism vitally depends of ketolysis, for their mitochondrial acetyl CoA supply. It is thus “a priori” not indicated to follow a ketogenic diet. It was then necessary to find out the clinical circumstances that introduced this diet. The recent publication of Klement [21] on the forgotten contribution of Wilhelm Brünings, is in this respect particularly interesting. Following the discoveries of Warburg on tumor metabolism [22, 23] (high glucose fermentation to lactate, in presence of oxygen). Brünings proposed to decrease the glucose supply to tumors, using a low carbohydrate diet and insulin induced hypoglycemia. However, it was necessary to feed patients with a diet replacing the carbohydrates, by lipids and proteins (the ketogenic diet was developed). The preliminary observations were encouraging; however, after several weeks there was a rebound of the tumors and much disappointment (See the detailed description by Klement [21].
What can we say on these trials? Evidently, in Diabetes type I the loss of beta cells and decrease of insulin release leads to hyperglycemia, with a loss of control over alpha cells that release glucagon, which elicits a ketogenic metabolism classically found in Diabetes. On the contrary, when one injects insulin to an individual, there is a drop of blood glucose, since tissues take it up. Here, insulin counteracts the release of catabolic glucagon that normally elicits the production of glucose and ketone bodies. Hence, in Burning’s trial, the initial effects of insulin-induced hypoglycemia must have been associated to a parallel decrease of ketogenesis, since insulin inhibits the alpha cell, via a mechanism that is not in this case GABA-mediated (we describe it later). The decrease of ketone bodies provoked by the insulin-mediated blockade of glucagon release in this initial phase of the trial coincides with the decrease of tumor sizes. Indeed, without ketone bodies, tumor cells could not make their mitochondrial acetyl CoA. However, with the gradual onset of the ketogenic diet, the supply of ketone bodies to the tumor starts again, and a rebound of the tumor sizes took place; the results of trial were disappointing.
The direct effect of insulin, over pancreatic cells has been recently unraveled [24]. Apparently, insulin acts on delta cells insulin receptors, eliciting a glucose influx through glucose transporters and through the activation of an electrogenic co- transporter of glucose and sodium (SGLT2), the sodium-glucose luminal transporter 2. The resultant increase of glycolysis forms ATP, which closes K ATP channels. The overall membrane depolarization then opens calcium channels and triggers an influx of calcium, which induces via ryanodine receptors, a further mobilization of calcium stores from the reticulum, activating the release of somatostatin from delta cells. The paracrine effect of somatostatin over neighboring cells is to inhibit both glucagon release from alpha cells and insulin release from beta cells. Thus, in relation to Brüning’s trial, injected insulin, which elicits hypoglycemia, will directly stimulate delta cells releasing somatostatin, which in turn inhibits the release of glucagon from alpha cells, the counter regulation by glucagon is OFF; neither glucose production nor ketogenesis take place after the insulin injection. Hence, the ketone bodies will decrease, depriving tumor cells of their vital acetyl CoA supply, in this initial phase of the trial tumor sizes decrease. Later, the ketogenic diet gradually provides to tumors ketone bodies, and ketolysis feeds their mitochondria with a vital acetyl CoA supply, while the rebound of tumors took place.
However, at an epigenetic level, beta hydroxybutyrate inhibits histone deacetylase (HDAC); acetylated histones increase, which may favor the expression of genes that are silent in cancer, such as P53, which could be favorable if it occurs. On the other hand, we know that the butyrate inhibition of HDAC induces the expression of embryonic genes; it is the case for fetal hemoglobin or utrophine and others. Thus, the expression embryonic M2 pyruvate kinase in tumor cells, instead of the M1 regulated adult form, might be a consequence of an increased supply of ketone bodies and HDAC inhibition, and this is not favorable, since the M2 form of PK, causes the glycolytic bottle neck in tumor cells.
SCOT inhibition: A possible cancer treatment
The therapeutic elements we propose, in relation to this metabolic presentation, have still to go through tests on animal models, before adding them to actual therapies that have a long experimental background. Moreover, even if the compounds proposed come from published observations, they need an evaluation for the toxicity of mixtures, which could be different from the toxicity of each compound, prescribed for other indications.
In Figure 2, the numbers indicate a selection of enzymes to target. There are six points of attack that we have chosen. The first target number 1 in figure 2, is the ketolytic enzyme SCOT. The list of inhibitors found in the Brenda chemical database, contains many compounds that are difficult to use or probably toxic, such as dinitrophenyl acetate, or 2, 2 difluoro succinate [25] others could indeed be tested: desulfo CoA, desulfopanteteine, acetylimidazole, 3 sulfopropanoate, N- acetylcysteamine, citrate, iodine and more. We then found in the work of Picard and Jenks [26] on the active center of SCOT a particularly interesting compound: acetohydroxamic acid, which inhibits the enzyme-substrate complex. Acetohydroxamic acid is available as tablets under the name of lithostat, against kidney stones and bladder infections, caused by bacteria raising the ammonia level in urine. Salicylhydroxamic acid is also an interesting compound to test. Moreover, we found that a class of histone deacetylase inhibitors is hydroxamic acid derivatives, such as suberoylanilide hydroxamic acid (SAHA), or trichostatine, or vorinostat and others, they do display anticancer properties [27]. However, we find no mention for a possible inhibition of SCOT in this observation, but this would indeed explain the observed anticancer properties. The compounds we preferably select for inhibiting SCOT are acetohydroxamic acid, and derivatives such as SAHA, or vorinostat. The next target, number 2 in figure 2 aims to decrease the efflux of citrate from the mitochondria to the cytosol, where citrate hydrolysis by ATP citrate lyase gives back acetyl CoA and OAA. In previous works, lipoic acid (the co-factor of PDH) associated to the ATP citrate lyase inhibitor hydroxycitrate, gave good results on animal tumors [28]. We think that lipoic acid has in fact slowed down the citrate condensation reaction; by probably reducing NAD into NADH, which inhibits citrate synthetase. Presently, we suggest keeping hydroxycitrate; there are also other ATP citrate lyase inhibitors such as bempedoic acid [29]. We would again associate hydroxycitrate and lipoic acid as previously, if lipoic acid does not cause a reactivation of SCOT after its inhibition by acetohydroxamic acid; if this was the case, we would omit lipoic acid. The acetyl CoA produced by ATP citrate lyase in the cytosol follows the lipogenic pathway, while OAA pushes the transamination chain leading in fine to pyruvate and lactate. OAA converts to malate, shuttles in mitochondria and gives aspartate that enters in several pathways that are not the subject of the present work. At the end of the lipogenic pathway we find fatty acid synthetase (FAS), this target is our number 3 in the figure 2, inhibitors are: cerulenin, C75 or orlistat, we retain orlistat for example, this would maintain the intermediate concentration of malonyl CoA that closes the beta oxidation of fatty acid[30].
It is necessary to decrease the cytosolic synthesis of acetyl CoA by inhibiting acetyl CoA synthetase (ACS), this enzyme uses as substrate acetate taken-up by tumor cells; this target is number 4 in figure 2. Acetyl CoA synthetase belongs to a class of enzymes forming adenylate intermediates, similarly to acetoacetyl CoA synthetase (AACS), which is target number 6 in figure 2, along the cholesterol synthetizing pathway. Inhibitors of adenylate forming enzymes are sulfonyladenosines or celecoxib derivatives (AR12, AR14), or adenosine 5’- ethyl phosphate, we also find a quinoxaline derivative [31, 32, 33, 14]. Other simpler compounds, such allicine from garlic, inhibit acetyl CoA synthetase [34]. This is also the case of an active substance from cow milk, which could be orotic acid [35]. Orotic acid and allicin are available as tablets. In between acetyl CoA synthetase (target 4), and acetoacetyl CoA synthetase (target 6); a cytosolic thiolase operates, this target is number 5 in figure 2; Inhibitors of thiolase such as trimetazidine (vastarel) or 4- pentenoic acid are available but difficult to handle [36].
A compound such as dichloroacetate, which activates PDH by inhibiting PDH kinase, displays a parallel blockade of ketolysis [37]. In the context of the present work, this is particularly interesting, since dichloroacetate has known anti-tumor effects. In addition, dichloroacetate presumably inhibits the acetyl CoA synthetase, which feeds acetate in the lipogenic pathway. Finally, along the cholesterol synthesis pathway, statins classically inhibit 3-hydrox 3- methyl glutaryl CoA synthetase and cholesterol synthesis, this target is number 7 in figure 2. A first choice for blocking the tumor could be acetohydroxamic acid or a derivative, plus hydroxycitrate, allicine, or orotic acid and lipoic acid (if it does not reactivate SCOT). Dichloroacetate orlistat and the other enzyme inhibitors that we mention because they validate the mechanism discussed, have to go through toxicity tests before. We only recommend compounds that one prescribes for other medical indications with no side effects, after testing the mixture on animal models.
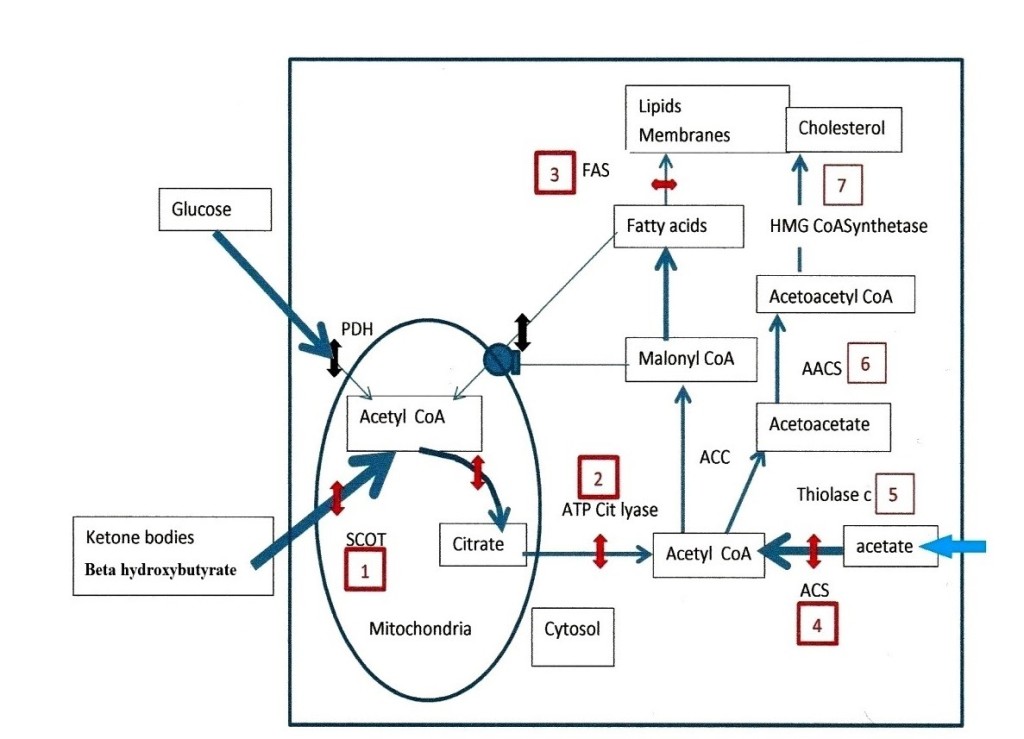
Figure 2.
Figure 2: Enzymatic targets to stop the progression of tumors. Glycolytic and fatty acid supplies of mitochondrial acetyl CoA are not operational in tumor cells: (Pyruvate dehydrogenase (PDH) is OFF and malonyl CoA inhibits the fatty acid carnityl transporter (double black interruptions). The only supply of acetyl CoA comes from ketolysis via succinyl-CoA: 3 oxoacid- CoA transferase (SCOT) target number 1, its inhibition by acetohydroxamic acid or derivatives is represented by the red double arrow, as for next targets. The citrate efflux and ATP citrate lyase is target 2, previously tested compounds are lipoic acid, and hydroxycitrate. At the end of the fatty acid synthesis pathway, target 3 is fatty acid synthetase (FAS), orlistat is a possible inhibitor but requires toxicity tests. Target 4 is acetyl CoA synthetase (ACS), inhibitors are allicine or orotic acid, others that we mention in the text such as sulfonyladenosines inhibit acetoacetylCoA synthetase (AACS) (target 6) they also require toxicity tests. In between, target 5 is a cytosolic thiolase, inhibitors are trimetazidine and 4 -pentenoic- acid they are difficult to handle. The last target is the enzyme 3 hydroxy 3-methyl glutaryl CoA synthetase (target 7) statins inhibit this enzyme and lower cholesterol but have side effects. In our opinion targets 1, 2 and 4 are those we would handle first, with acetohydroxamic, hydroxycitrate and allicine or orotic acid, we would add lipoic acid if it does not reactivate SCOT.
Discussion
By inhibiting SCOT and the ketolytic source of acetyl CoA, one suppresses the only supply of acetyl CoA for tumor cell mitochondria; indeed, the other sources glycolytic and fatty acid are not operating, since PK and PDH are OFF, forming the glycolytic bottle neck, while the fatty acids degradation is OFF, when fatty acid synthesis is ON. Hence, the blockade of SCOT deprives tumor cells of their vital acetyl CoA supply; they should then regress. In the cytosol of tumor cells, a supply of acetate can still feed acetyl CoA synthetase and the lipogenic pathway, inhibiting this entry as well, would make it difficult for tumor cells to synthesize their membranes, they should die or have to normalize their metabolism.
In earlier works, we proposed to try to normalize cancer metabolism, by controlling the starter mechanism that rewires metabolic pathways in cancer: the pancreatic GABA trigger, the elevated cAMP, the growth hormone – ATGL- DAG- PKC axis. The latter leads to the inhibition protein phosphatases and activation of kinases, and perturbs metabolic switches controlling cancer metabolism in differentiated cells and tumor cells (16). Normalizing cancer metabolism requires an elevated number of compounds. We found some active minimal combinations but much work remains unfinished, in spite of interesting possibilities to test. The present proposal is different, since it takes advantage of the weak point of tumor cell metabolism; that vitally depends of SCOT and acetyl CoA synthetase. Their inhibition should hold back the development of tumors, giving a new possibility to add to the treatment of cancer.
Footnotes
Conflict of interest statement: The authors have no conflict of interest related to the manuscript
Manuscript source: Invited manuscript.
References
- Solimena M, Folli F, Denis-Donni S, Comi GC, De Camilli P, et al. (1988) Autoantibodies to glutamic acid decarboxylase in a patient with stiff-man syndrome, epilepsy, and type I diabetes. N Engl J Med 318: 1012-20.
- Wan Y, Wang Q, Prud’homme GJ (2015) Gabaergic system in the endocrine pancreas: A new target for diabetes treatment. Diabetes Metabolic Syndrome and Obesity 8: 79-87.
- Wendt A, Blinir B, Buschard K, Gromada J, Salehi A, et al. (2004) Glucose inhibition of glucagon secretion from rat alpha-cells is mediated by GABA released from neighboring beta-cells. Diabetes 53:1038-45.
- Braun M, Wendt A, Buchard K, Salehi A, Sewing S, et al. (2004) GABA B receptor activation inhibits Exocytosis in rat pancreatic-beta- cells by G-protein-dependent activation of calcineurin. J Pysiol 559: 397-409.
- Taneera J, Jin Z, Jin Y, Muhammed S.J, Zhang E, et al. (2012) Aminobutyric acid (GABA) signaling in human pancreatic islets is altered in type 2 diabetes. Diabetologia 55: 1985-1994.
- Israël M (2012) A possible primary cause of cancer: deficient cellular interactions in endocrine pancreas. Molecular Cancer 11: 63-68.
- Israël M (2012) A primary cause of cancer: GABA deficiency in endocrine pancreas. Cancer Therapy 8: 171-183.
- Israël M (2014) Signaling and metabolism in cancer: Endocrine pancreas deficiency and hybrid anabolism-catabolism, drugs that undo the process. Cancer Therapy 10: 1-12.
- Israël M (2014) Comment on cancer metabolism and on the role of the endocrine pancreas. J Clin Med Res 6: 490-491.
- Israël M (2017) Altered Controls Transforming Normal Metabolism into Carcinogenic. J Cancer Prev Curr Res 700228-233.
- Mazurek S, Eigenbrodt E (2003) The tumor metabolome. Anticancer Res 23: 1149-54.
- Eigenbrodt E, Gerbracht U, Mazurek S, Presek P, Friis R. Carbohydrate metabolism and neoplasia: New perspectives for diagnosis and therapy. In Biochemical and Molecular Aspects of Selected Cancers. Prestlow TG, Prestlow TP, editors, Academic Press Inc (1994): 311-385.
- Eto M (2009) Regulation of cellular protein phosphatase 1 (PP1) by phosphorylation of the CPI-17 family, C- kinase- activated PP1 inhibitors. J Biol Chem 284: 35273-7.
- Mayer N, Schweiger M, Romauch M, Eichmann TO, et al. (2013) Development of small-molecule inhibitors targeting adiposetriglyceride lipase. Nat Chem Biol 9: 785-7.
- Israël M, Schwartz L (2011) The metabolic advantage of tumor cells. Molecular Cancer 10: 1-12.
- Israël M (2019) Metabolic rewiring of stem cells and differentiated cells in cancer: the hypothetical consequences of a GABA deficiency in endocrine pancreas. J Cancer Metastasis Treat: 5-12.
- Comerford SA, Huang Z, Du X, Wang Y, Cai L, et.al. (2014) Acetate dependence of tumors. Cell 159:1591-602.
- Israël M, Tucek S (1974) Utilisation of acetate and pyruvate for the synthesis of ‘total’, ‘bound’ and ‘free’ acetylcholine in the organ of Torpedo. J Neurochem 22: 487-91.
- Martinez-Outschoorn UE, Lin Z, Whitaker-Menezes D, Howell A, Sotgia F, et al. (2012) Ketone body utilization drives tumor growth and metastasis. Cell Cycle 11: 3964-3971.
- Zhang S, Xie C (2017) The role of OXCT1 in the pathogenesis of cancer as a rate-limiting enzyme of ketone body metabolism. Life Sciences 183:110-115.
- Klement RJ (2019) Wilhelm Brünings’ forgotten contribution to the metabolic treatment of cancer utilizing hypoglycemia and very low carbohydrate (ketogenic) diet. Journal of traditional and Complementary Medicine 9: 192-200.
- Warburg O (1956) On the origin of cancer cell. Science 123:309-14.
- Warburg O (1956) on respiratory impairment in cancer cells. Science 124: 269-70.
- Vergari E, Knudsen JG, Ramracheya R, Salehi A, Zhang Q, et al. (2019) Insulin inhibits glucagon release by SGTL2- induced stimulation of somatostatin secretion. Nat Commun 10: 139-50.
- Fenselau A, Wallis K (1974) Substrate specificity and mechanism of action of acetoacetate Coenzyme A transferase from rat heart. Biochemistry 13: 3884-3888.
- Pickart CM, Jencks WP (1979) Formation of stable anhydrides from CoA transferase and hydroxamic acids. The Journal of Biological Chemistry 254: 9120-9129.
- Pal D, Saha S (2012) Hydroxamic acid –A novel molecule for anticancer therapy. J Adv Pharm Technol Res 3: 92-98.
- Schwartz L, Abolhassani M, Guais A, Sanders E, Steyaert JM, et al. (2010) A combination of alpha lipoic acid and calcium hydroxycitrate is efficient against mouse cancer models: preliminary results. Oncol Rep 23: 1407-16.
- Pinkosky SL, Newton RS, Day EA, Ford RJ, Austin RC, et al. (2016) Liver-specific ATP-citrate lyase inhibition by bempedoic acid decrease LDL-C and attenuates atherosclerosis. Nat Commun 7: 13457-70.
- Flavin R, Peluso S, Nguyen PL, Loda M (2010) Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol 6: 551-562.
- Lux MC, Standke LC, Tan DS (2019) Targeting adenylate-forming enzymes with designed sulfonyladenosine inhibitors. J antibiot (Tokyo) 72: 325-349.
- Koselny K, Green J, Favazzo L, Glazier VE, Didone L, et al. (2016) Antitumor/Antifungal Celecoxib derivative AR-12 is a non- nucleoside inhibitor of ANL- family adenylating enzyme acetyl CoA synthetase ACS. Infectious Diseases 2: 268-280.
- Grayson NA, Westkaemper RB (1988) Stable analogs of acyl adenylates, inhibition of acetyl- and acyl-CoA synthetase by 5’-alkylphosphate. Life Sci 43: 437-44.
- Focke M, Feld A, Lichtenthaler HK (1990) Allicin, a naturally occuring antibiotic from garlic, specifically inhibits acetyl-CoA Synthetase. FEBS 261: 106-108.
- Bernstein BA, Richardson T, Amundsen CH (1977) Inhibition of Cholesterol biosynthesis and Acetyl-CoA synthetase by bovine Milk and Orotic acid. Journal of Dairy Science 60: 1846-1853.
- Schulz H (1983) Metabolism of 4-pentenoic acid and inhibition of thiolase by metabolites of 4-pentenoic acid. Biochemistry 22: 1827-32.
- Ayat M (2018) Dichloroacetate is promising for treating hematological malignancy through inhibiting ketone bodies oxidation: Towards better understanding of its anticancer mechanisms. American Journal of Cancer Prevention 6: 5-8.