DOI: 10.31038/MGJ.2022512
Abstract
The complete mitochondrial genome (mitogenome) of the comma butterfly, Polygonia c-aureum (Lepidoptera: Nymphalidae) is determined in this study. It is a circular molecule of 15,208 bp, containing 13 protein-coding genes, 2 ribosomal RNA (rRNA) genes, 22 transfer RNA (tRNA) genes, and an A+T-rich region, which is a common feature of lepidopteran mitogenomes. Based on nucleotide sequences of 13 protein-coding genes, we reconstructed the phylogenetic relationships among 87 species of the family Nymphalidae using Bayesian Inference (BI) and Maximum Likelihood (ML) methods, and calculated the divergence times using multiple fossil calibrations. The phylogenetic analyses supported the sister-group relationship between the subfamilies Nymphalinae and Cyrestinae. Moreover, monophyly of the Nymphalidae was strongly supported. The results were highly consistent with the traditional relationships within the Nymphalidae from morphological data. For the first time, our results suggest that the genus Polygonia diverged from the common ancestor of the rest of Nymphalinae at 45.64 Ma. In addition, the first divergence time in the Nymphalidae is in the Early Cretaceous, about 89.72 Ma.
Keywords
Mitochondrial genome, Molecular phylogeny, Divergence time, Nymphalidae, Polygonia c-aureum
Introduction
The Nymphalidae is the largest family of butterflies, including 7,200 species belonging to 600 genera and 12 subfamilies [1-6]. Consequently, it has been the subject of intense studies [7-9]. Nymphalidae is the first taxa that helped us to begin to understand the complex relationships between insects and their host plants [10], the effects of habitat fragmentation on the population dynamics of endangered species [11], and the genetic mechanisms behind the developmental pathways of morphological features [12], and the coevolutionary interactions between organisms in mimicry rings and aposematic coloration [13,14]. Especially the butterflies of the subfamily Nymphalinae [5] have extensively contributed to our knowledge on ecological and evolutionary processes [15-18]. However, the phylogenetic relationships among the different subfamilies and tribes have been chaotic because of the variable shapes and life cycles, it made them become the argue focus for taxonmists [6,19-21]. There are still several competing classification schemes based on different data sets and researchers [5,21,22]. With the development of sequencing technologies and increasing number of molecular data set, more and more researches investigated the phylogenetic relationships of butterflies. For example, [23] using the wingless gene, and [5] using COI, EF-1α and wingless genes, both including good taxonomic coverage of the Nymphalidae, showed that many of the traditional subgroups are monophyletic. [7] inferred a robust phylogenetic hypothesis based on 10 genes and 235 morphological characters.
Meanwhile, there are many difficulities in the research of orgin and evolution in most of the families, in view of the lack of fossils data. [7] used a surprisingly good fossil record for the Nymphalinae to estimate the ages of diversification major lineages using Bayesian relaxed clock methods, suggesting that the age of Nymphalidae is older than 70 million years. [24] explored the divergence time in butterflies using the sequences of ultraviolet-sensitive (UVRh), blue-sensitive (BRh), long-wavelength sensitive (LWRh) opsins, EF-1α and COI obtained from 27 taxa representing the five major butterfly families.
The comma butterfly, Polygonia c-aureum, is a major defoliator leaf pest on the scandent hostlant Humulus scandens (Lour.) Merr., which is used for medicine in China [25]. Here, we sequenced the complete mitochondrial genome (mitogenome), which could can be used to develop molecular markers for phylogenetics and, identification, and also to examine the evolution of Nymphalidae. In addition, we hope our study would be useful for the prevention and control of insect pests.
In this study, based on the complete mitogenome sequences of P. c-aureumy and additional homologous sequences of 86 species downloaded from GenBank, we estimated the divergence times of Nymphalidae, to enhance our understanding of the origin and evolution of this family, and to provide a relative accurate results for estimating divergence times of butterflies.
Materials and Methods
Sample Collection and DNA Extraction
The adult specimens of P. c-aureum were collected from Nanjing, Jiangsu Province in China. After an examination of external morphology for identification, the fresh adult specimens were directly frozen and maintained at -80°C until DNA extraction. Total genomic DNA was extracted from adult butterfly tissues, typically thorax or abdomen, using the Wizard Genomic DNA Purification Kit (Promega, Beijing, China) according to the manufacturer’s instruction. The extracted DNA was stored at -20°C and used for PCR amplification of the complete mitogenome.
Primers Design, PCR Amplification and DNA Sequencing
In order to amplify the complete mitogenome of P. c-aureum, nineteen pairs primers were designed and synthesized. Among them, four pairs are lepidoptera universal primers [26,27], twelve pairs specific primers for this study were designed using Primer Premier 5.0 software [28] and the remainder of three pairs primers were the combination of universal primers and specific primers. Detailed information about primers used in this study are shown in Table 1.
Table 1: Primers used for amplification of the Polygonia c-aureum mitogenome
| Fragment | Region | Primer (J/N) |
Primer sequence (J/N) 5’→3’ |
| P1 | ND2 | N2-J1c/N2-N1c | ATAAGCTAAATAAGCTTTTGGGTTCATA/ATTATTAATGCAGATAATATTCATCCTAAATT |
| P2 | ND2—COI | J-556c/N-2904c | AATAGGATCAGCACCAT/CAAGAAATGTTGAGGGA |
| P3 | COI—COII | C1-J-2167a/N-3649a | TTGATTTTTCGGACATCCTGAAGT/CCGCAAATTTCTGAACATTGACCA |
| P4 | COII—ATP8 | J-3241c/N-3849c | TTGATTTTTCGGACATCCTGAAGT/CCGCAAATTTCTGAACATTGACCA |
| P5 | COII—ATP6 | J-3455c/N4734c | TATTGCACTCCCATCCC/GTTCTTCTAAGGAGGGT |
| P6 | ATP6 | C2-Jc/C3-Nc | ATTTGTGGAGCTAATCATAG/GGTCAGGGACTATAATCTAC |
| P7 | ATP6—COIII | J-4556c/N-5346c | TTACCCTCCTTAGAAGAACA/AAATGTCGGATAAAGCAAGT |
| P8 | COIII—ND3 | C2-J-3696a/N3-N-5731a | GAAATTTGTGGAGCAAATCATAG/TTTGGATCAAACCCACATTC |
| P9 | ND3—ND5 | C3-N5-5407c/N5-N-7793b | GCTGCAGCTTGATATTGACA/TTGGGTTGGGATGGTTTAGG |
| P10 | ND5—ND4 | N5-J-7572a/N4-N-9153a | AAAAGGAATTTGAGCTCTTTTAGT/TGAGGTTATCAACCAGAGCG |
| P11 | ND4—Cytb | N4-J-8502a/CB-N-11328a | GTAGGAGGAGCTGCTATATTAG/GGCAAATAGGAAATATCATTC |
| P12 | ND4—Cytb | N4-J2c/CB-N2c | CCCTAATAATAAAGGCAATG/TTATCAACAGCAAATCCACC |
| P13 | Cytb | CB-J-10933a/N-11526c | GTTTTACCATGAGGTCAAATATC/TTCTACAGGTCGGGCTCCGATTCA |
| P14 | Cytb—ND1 | J-11338c/N-12051c | CATATTAAACCCGAATGATATTT/GTATTTGCTGAAGGTGAATCAGA |
| P15 | ND1—16S | N1-16S-J11876b/N-13000c | CGAGGTAAAGTACCACGAACTCA/TTACCTTAGGGATAACAGCGTAA |
| P16 | 16S | J-12609c/N-13554c | ACCATTACATTTATCTGCCA/ATTTTAGGGGATAAGCTTTA |
| P17 | 16S | J-13310c/N-14094c | ATCAGGGGGCAGATTAAACTTTAA/CTAGAAAGATCAAATTAGAGCT |
| P18 | 16S—12S | J-13653c/N-14360c | CGATTAACATTTCATTTC/ATTGATAATCCACGAAT |
| P19 | 12S—ND2 | 12S-N2-Jc/N2-Nc | CTCTACTTTGTTACGACTTATT/TCTAGGCCAATTCAACAACC |
a: Primers modified from Simon et al. (1994) up to this mtgenome
b: Primers from Simon et al. (2006)
c: Primers newly designed for this genome
Some PCR reactions (the target fragments <2 kb) were performed in a 25 μL volume with 0.2 μL rTaq (TaKaRa Co., Dalian, China), 1 μL of DNA, 2.0 μL dNTPs, 2.0 μL 25 mM MgCl2, 2.5 μL 10× rTaq buffer (Mg2+ free), 0.5 μL each primer and 16.3 μL sterile distilled H2O. The PCR amplification was performed using the following cycling protocol: an initial denaturation for 5 min at 94°C, followed by 35 cycles of denaturation at 94°C for 30 seconds, annealing at 50°C~59°C (depending on primer pairs) for 30 seconds, extension at 72°C for 1~2 min, with a subsequent 10 min final extension at 72°C. Besides, the other PCR reactions (the target fragments ≥ 2 kb) were carried out with 25 μL reaction volume containing 0.2 μL of LATaq (TaKaRa Co., Dalian, China), 1 μL of DNA, 4.0 μL dNTPs, 2.5 μL 10×Taq buffer (Mg2+ plus), 16.3 μL sterile distilled H2O and 0.5 μL each primer. The fragments were amplified under the following cycling protocol: 5 min of initial denaturation at 94°C, followed by 35 cycles of denaturation for 30 seconds at 94°C, annealing at 50°C~59°C (depending on primer pairs) for 30 seconds, extension at 72°C for 1~2 min, with additional 5 seconds for each cycle, and a final extension for 10 min at 72°C.
Products were examined by electrophoresis on 1% agarose gel. All the PCR fragments were directly sequenced from both strands by Jin Si Rui Company, Nanjing, China and Sheng Gong Company, Shanghai, China with the PCR primers.
Sequence Assembling and Annotation
The raw sequences files were proofread and assembled manually using the SeqMan module of the Lasergene 8.0 software (DNASTAR, Madison, WI, USA) [29]. The probable locations of the sequences were confirmed by BLAST search function on the NCBI website and comparison with the other lepidopteran sequences which can be obtained in GenBank. By using MEGA7.0, we determined the translation of 13 PCGs open reading frames [30]. The base composition of nucleotide sequences was described by skewness and measured according to the formulas (AT skew = [A−T]/[A+T], GC skew = [G−C]/[G+C]) [31]. 22 tRNA were confirmed using the program tRNAscan-SE. The proposed cloverleaf secondary structures within these tRNA genes and anticodon sequences were calculated using the tRNAscan-SE Search Server available online (http://lowelab.ucsc.edu/tRNAscan-SE/) [32]. We drew the secondary structure of tRNA by using the RNA structure program DNASIS MAX v.3.0 [33]. The secondary structure of the tRNASer (AGN) was developed as proposed by [34]. Annotation was checked by comparison with tRNA determined for other lepidopteran species. Ribosomal RNA genes (rRNAs) were identified by NCBI Internet BLAST search.
Phylogenetic Analyses
To further probe into the phylogenetic relationship of Nymphalidae, a total of 84 complete mitogenomes and three uncomplete mitogenomes were chosen for the phylogenetic analyses based on the concatenated set of amino acid from 13 protein coding genes. The GenBank accession numbers used in this study were listed in Table 2. Among the 87 species, Coreana raphaelis (DQ102703.1), Japonica lutea (KM655768.1), Eurema hecabe (KC257480.1), Colias erate (KP715146.1), Curetis bulis (JX262888.1), Papilio bianor (KF859738.1), P. machaon (HM243594.1) and Leptidea morsei (JX274648.1) were selected as outgroups (Table 2). The PCG sequences of 87 species were aligned by using MEGA7.0 [30]. Sites with more than 90% gaps were excluded from the analysis. We chose two analysis approaches, Bayesian Inference (BI) and Maximum Likelihood (ML) to reconstruct phylogenetic relationships. We used the MrModeltest 2.3 [35] to select the best model for the ML and BI analyses. Thirteen datasets were established to calculate the best model for each PCG. According to the Akaike information criterion, the GTR + G model was selected as the most model appropriate for ND4L, and the GTR+I+G model was selected for other genes. The BI analysis was performed using MrBayes vers. 3.1.2 [36] under both of the models. The analysis were run twice simultaneously for 10,000,000 generations with every 1000 trees sampled. We discarded the first 1000,000 generations (1000 samples) as burn-in (based on visual inspection of the convergence and stability of the log likelihood values of the two independent runs). The ML analysis were performed using the program MEGA7.0 [30] with the same model. The bootstrap analysis were performed with 1000 replicates. Resulting tree files were inspected in FigTree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/).
Table 2: Taxonomy, GenBank accession numbers, and mitogenome sizes of 87 the mitochondrial genomes used for the phylogenetic analysis, sourced from GenBank databases.
|
Subfamily |
Species | Genome
size (bp) |
GenBank accession no. |
| Nymphalinae | Polygonia c-aureum | 15208 | KX096653 |
| Inachis io | 15250 | KM592970.1 | |
| Junonia orithya | 15214 | KF199862.1 | |
| Yoma sabina | 15330 | KF590535.1 | |
| Hypolimnas bolina | 15260 | KF990127.1 | |
| Melitaea cinxia | 15170 | GQ398377.1 | |
| Kallima inachus | 15150 | HM243591.1 | |
| Cyrestinae | Cyrestis thyodamas | 15254 | KF990125.1 |
| Dichorragia nesimachus | 14367 | KF990126.1 | |
| Biblidinae | Ariadne ariadne | 15179 | KF990123.1 |
| Hamadryas epinome | 15207 | KM378244.1 | |
| Apaturinae | Sasakia charonda | 15244 | AP011824.1 |
| Sasakia charonda kuriyamaensis | 15222 | AP011825.1 | |
| Sasakia funebris | 15233 | JX131328.1 | |
| Euripus nyctelius | 15417 | KR020515.1 | |
| Apatura ilia | 15242 | JF437925.1 | |
| Apatura metis | 15236 | JF801742.1 | |
| Timelaea maculata | 15178 | KC572131.1 | |
| Chitoria ulupi | 15279 | KP284544.1 | |
| Limenitidinae | Athyma kasa | 15230 | KF590524.1 |
| Athyma cama | 15269 | KF590526.1 | |
| Athyma perius | 15277 | KF590528.1 | |
| Athyma opalina | 15240 | KF590551.1 | |
| Athyma selenophora | 15208 | KF590529.1 | |
| Pandita sinope | 15257 | KF590530.1 | |
| Athyma sulpitia | 15268 | JQ347260.1 | |
| Parasarpa dudu | 15236 | KF590537.1 | |
| Athyma asura | 15181 | KF590542.1 | |
| Abrota ganga | 15356 | KF590536.1 | |
| Lexias dirtea | 15250 | KF590531.1 | |
| Tanaecia julii | 15316 | KF590548.1 | |
| Dophla evelina | 15320 | KF590532.1 | |
| Euthalia irrubescens | 15365 | KF590527.1 | |
| Neptis philyra | 15164 | KF590552.1 | |
| Neptis clinia | 15189 | KM244664.1 | |
| Neptis soma | 15130 | KF590533.1 | |
| Pantoporia hordonia | 15603 | KF590534.1 | |
| Bhagadatta austenia | 15615 | KF590545.1 | |
| Parthenos sylvia | 15249 | KF590550.1 | |
| Heliconiinae | Fabriciana nerippe | 15140 | JF504707.1 |
| Argynnis paphia | 15208 | KM592975.1 | |
| Argynnis hyperbius | 15156 | JF439070.1 | |
| Argynnis childreni | 15131 | KF590547.1 | |
| Issoria lathonia | 15172 | HM243590.1 | |
| Cethosia biblis | 15286 | KR066948.1 | |
| Acraea issoria | 15245 | GQ376195.1 | |
| Heliconius pachinus | 15369 | KM014809.1 | |
| Heliconius cydno | 15367 | KM208636.1 | |
| Heliconius melpomene rosina | 15327 | KP100653.1 | |
| Heliconius melpomene | 15328 | HE579083.1 | |
| Heliconius ismenius | 15346 | KP294327.1 | |
| Heliconius hecale | 15338 | KM068091.1 | |
| Heliconius clysonymus | 15302 | KP784455.1 | |
| Heliconius sara | 15372 | KP281778.1 | |
| Satyrinae | Stichophthalma louisa | 15721 | KP247523.1 |
| Elymnias hypermnestra | 15167 | KF906484.1 | |
| Triphysa phryne | 15143 | KF906487.1 | |
| Lethe dura | 15259 | KF906485.1 | |
| Mycalesis mineus | 15267 | KM244676.1 | |
| Neope pulaha | 15209 | KF590543.1 | |
| Ninguta schrenckii | 15261 | KF881052.1 | |
| Pararge aegeria aegeria | 15240 | KJ547676.1 | |
| Callerebia suroia | 15208 | KF906483.1 | |
| Hipparchia autonoe | 15489 | GQ868707.1 | |
| Melanargia asiatica | 15142 | KF906486.1 | |
| Ypthima akragas | 15227 | KF590553.1 | |
| Melanitis phedima | 15142 | KF590538.1 | |
| Melanitis leda | 15122 | JF905446.1 | |
| Charaxinae | Polyura arja | 15363 | KF590540.1 |
| Polyura nepenthes | 15333 | KF990128.1 | |
| Calinaginae | Calinaga davidis | 15267 | HQ658143.1 |
| Danainae | Danaus plexippus | 15314 | KC836923.1 |
| Danaus chrysippus | 15236 | KF690637.1 | |
| Tirumala limniace | 15285 | KJ784473.1 | |
| Parantica sita | 15211 | KF590544.1 | |
| Ideopsis similis | 15200 | KJ476729.1 | |
| Euploea midamus | 15187 | KJ866207.1 | |
| Euploea mulciber | 15166 | HQ378507.1 | |
| Libytheinae | Libythea celtis | 15164 | HQ378508.1 |
| Out groups
Theclinae |
Coreana raphaelis |
15314 |
DQ102703.1 |
| Japonica lutea | 15225 | KM655768.1 | |
| Curetinae | Curetis bulis | 15162 | JX262888.1 |
| Papilioninae | Papilio bianor | 15357 | KF859738.1 |
| Papilio machaon | 15185 | HM243594.1 | |
| Dismorphiinae | Leptidea morsei | 15122 | JX274648.1 |
| Coliadinae | Eurema hecabe | 15160 | KC257480.1 |
| Colias erate | 15184 | KP715146.1 |
Divergence Time Estimation
The analyses were performed based on sequences of 13 PCGs from 87 species, including eight outgroups. The program BEAST 2 [37] was used to estimate divergence times, with calibrations using five fossils nodes. Three fossils of Vanessa amerindica, Prodryas persephone and Lithopsyche styx were found in the Florissant formation in Colorado, which were formed in the early Oligocene and were thought to be related to the extant genus Hypanartia about 34 Ma in age. The fourth fossil is a hind wing that has been assigned to the extant genus Aglais, which was found in the Karagan deposits from the Miocene and has been dated at 14 Ma [38]. The last fossil is Dynamine alexaen deposits from the Miocene [39]. In addition, we used the results from Wahlberg et al. as secondary calibration point to calibrate the age of the first split in Nymphalidae at 90 Ma [7] and Papilionoidea at 104 Ma [40]. According to the result of our study, the Bayesian relaxed clock analyses were carried out with the program BEAST 2 [37]. The XML file for the beast analysis was created using BEAUti (in the BEAST package) with the following non-default settings and priors: the site model was set to the GTR +Γ distribution with default parameters, the clock model was set to a relaxed clock with uncorrelated rates, the tree model was set to a Yule process of speciation. The Markov chain Monte Carlo (MCMC) analyses were run for 100 million generations, sampling every 2000 generations and the first 25% discarded as burn-in. We used Tracer v1.5 to assess whether the likelihood traces of the four runs had converged to a stable equilibrium and that ESS values were above 200 for all parameters.
Results and Discussion
Genome Organization, Gene Arrangement, and Base Composition
The mitogenome of P. c-aureum (GenBank accession no. KX096653) is a closed circular molecule of 15,208 bp in size and similar to a typical insect mitogenome. The organization of the skipper mitogenome was shown in Figure 1. It contains the complete set of 37 genes, including 13 protein-coding genes (ND1-6, ND4L, COI-III, Cytb, ATP6, ATP8), 2 rRNA genes (12S and 16S), 22 putative tRNA genes, and an A+T-rich region (Figure 1). Similar to many insect mitogenomes, the majority (J) strand encodes more genes (9 PCGs and 14 tRNAs), whereas the minority (N) strand encodes lesser genes (4 PCGs, 8 tRNAs and 2 rRNAs) (Table 3). The order of genes and the orientation of the mitogenome of P. c-aureum are consistent with those sequenced lepidopteran mitogenomes. The nucleotide composition of the mitogenome of P. c-aureum is: A = 40.08%, T = 40.56%, G = 7.44% and C = 11.92% (Table 4). A + T content is 80.64%. Like other lepidopterans, the nucleotide composition of the P. c-aureum mitogenome is also biased toward A or T. This value is well in the range of the lepidopteran mitogenome, from 77.84 to 82.66%, which show a remarkable variability. Nucleotide skew statistics for the complete majority strand of P. c-aureum is AT-skew = −0.06 and GC-skew = −0.23 (Table 4), indicating slight A or T skews. A similar trend has been observed in many previously sequenced lepidopteran mitogenomes that the value of AT-skew varies from −0.031 (Eriogyna pyretorum) to 0.059 (Bombyx mori) and the GC-skew is always negative ranging from −0.318 (Ochrogaster lunifer) to −0.178 (Adoxophyes honmai) [41].
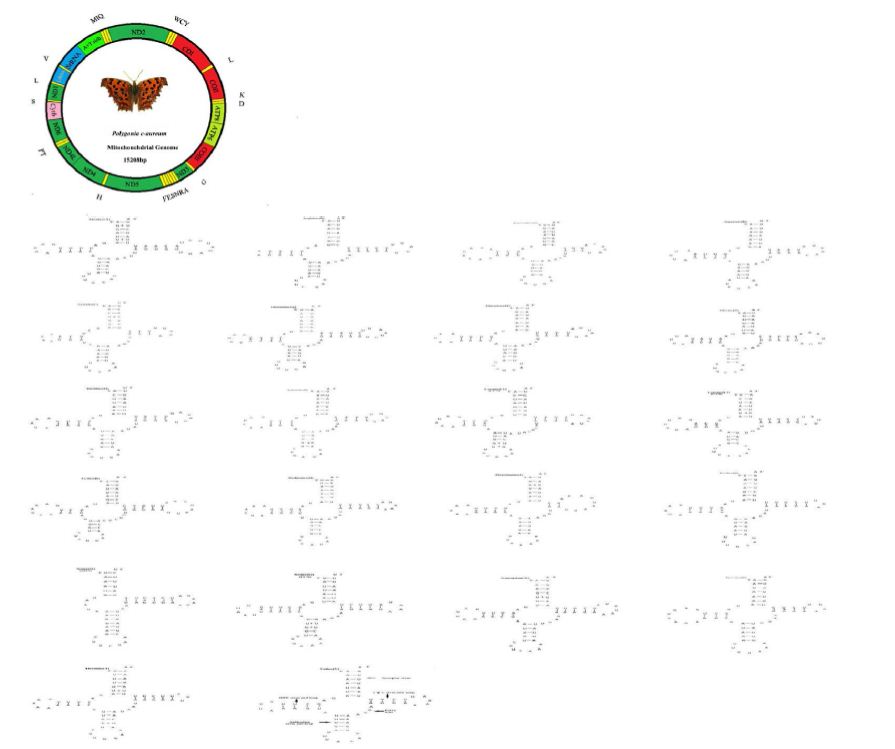
Figure 1: Map of the circular mitochondrial genome of Polygonia c-aureum. Different colors represent different regions. The abbreviations for the genes are as follows: COI-III stands for cytochrome oxidase subunits, Cytb for cytochrome b, and ND1-6 for NADH dehydrogenase Components. tRNAs are indicated by one-letter symbol according to the IUPAC-IUB single letter amino acid codes
Table 3: Annotation and gene organization of the Polygonia c-aureum mitogenome. Strands of the genes are presented as J for majority and N for minority strand. IN, negative numbers indicate that adjacent genes overlap, positive numbers indicate that intergenic sequences
|
Gene |
Strand | Nucleotide no. | Size(bp) | IN | Anticodon | Start codon |
Stop codon |
| tRNAMet |
J |
1-68 | 68 | 0 | CAT | – |
– |
|
tRNAIle |
J | 69-133 | 65 | 1 | GAT | – |
– |
| tRNAGln |
N |
135-203 | 69 | 46 | TTG | – |
– |
| ND2 |
J |
250-1263 | 1014 | -2 | – | ATT |
TAA |
| TrnaTrp |
J |
1262-1330 | 69 | -8 | TCA | – |
– |
| tRNACys |
N |
1323-1384 | 62 | -1 | GCA | – |
– |
| tRNATyr |
N |
1384-1448 | 65 | 4 | GTA | – |
– |
| COI |
J |
1453-2983 | 1531 | 0 | – | CGA |
T– |
| tRNALeu(UUR) |
J |
2984-3050 | 67 | 0 | TAA | – |
– |
| COII |
J |
3051-3726 | 676 | 0 | – | ATG |
T– |
| tRNALys |
J |
3727-3797 | 71 | -1 | CTT | – |
– |
| tRNAAsp |
J |
3797-3862 | 66 | 0 | GTC | – |
– |
| ATP8 |
J |
3863-4036 | 174 | -7 | – | ATT |
TAA |
| ATP6 |
J |
4030-4707 | 678 | -1 | – | ATG |
TAA |
| COIII |
J |
4707-5495 | 789 | 2 | – | ATG |
TAA |
| tRNAGly |
J |
5498-5566 | 69 | -3 | TCC | – |
– |
| ND3 |
J |
5564-5920 | 357 | 0 | – | ATA |
TAA |
| tRNAAla |
J |
5921-5991 | 71 | -1 | TGC | – |
– |
| tRNAArg |
J |
5991-6055 | 65 | 0 | TCG | – |
– |
| tRNAAsn |
J |
6056-6121 | 66 | 2 | GTT | – |
– |
| tRNASer(AGN) |
J |
6120-6179 | 60 | 9 | GCT | – |
– |
| tRNAGlu |
J |
6189-6254 | 65 | 10 | TTC | – |
– |
| tRNAPhe |
N |
6265-6329 | 65 | -2 | GAA | – |
– |
| ND5 |
N |
6328-8061 | 1734 | 0 | – | ATT |
TAT |
| tRNAHis |
N |
8062-8127 | 66 | -1 | GTG | – |
– |
| ND4 |
N |
8127-9466 | 1340 | 3 | – | ATG |
TA- |
| ND4L |
N |
9470-9757 | 288 | 2 | – | ATG |
TAA |
| tRNAThr |
J |
9760-9824 | 65 | 0 | TGT | – |
– |
| tRNAPro |
N |
9825-9889 | 65 | 2 | TGG | – |
– |
| ND6 |
J |
9992-10419 | 528 | 16 | – | ATT |
TAA |
| Cytb |
J |
10436-11587 | 1152 | 0 | – | ATG |
TAA |
| tRNASer(UCN) |
J |
11588-11655 | 68 | 20 | TGA | – |
– |
| ND1 |
N |
11676-12614 | 939 | 1 | – | ATG |
TAT |
| tRNALeu(CUN) |
N |
12616-12684 | 69 | -1 | TAG | – |
– |
| 16S |
N |
12684-14019 | 1336 | -1 | – | – |
– |
| tRNAVal |
N |
14019-14082 | 64 | 0 | TAC | – |
– |
| 12S |
N |
14083-14857 | 775 | 0 | – | – |
– |
| A+T-rich |
– |
14858-15208 | 388 | – | – | – |
– |
Table 4: Composition and skewness of Polygonia c-aureum mitogenome regions. # = position
|
Nt |
Whole mtDNA | PCG | rRNAs | tRNAs | ||
| 1st# | 2nd# |
3rd# |
||||
| A % |
40.08 |
30.93 | 33.89 | 35.60 | 39.74 |
40.73 |
| T % |
40.55 |
47.43 | 46.53 | 43.42 | 45.00 |
40.25 |
| C % |
11.92 |
10.71 | 9.43 | 10.21 | 10.18 |
10.88 |
| G % |
7.44 |
10.93 | 10.15 | 10.77 | 5.07 |
8.15 |
| A+T % |
80.64 |
78.36 | 80.42 | 79.02 | 84.75 |
80.97 |
| C+G % |
19.36 |
21.64 | 19.58 | 20.98 | 15.25 |
19.03 |
| AT-Skew |
-0.0058 |
-0.2105 | -0.1572 | -0.0990 | -0.062 |
0.006 |
| GC-Skew |
-0.2314 |
0.0099 | 0.0369 | 0.0268 | -0.335 |
-0.144 |
Protein-coding Genes
The PCGs of the P. c-aureum mitogenome include 7 NADH dehydrogenase subunits, 3 cytochrome c oxidase subunits, 2 ATPase subunits, and one cytochrome b gene. The PCGs of the mitogenome consists of 3,715 codons in total, except the termination codons. The start and stop codons of the 13 PCGs in the P. c-aureum mitogenome are shown in Table 3. Seven PCGs share the start codon ATG (COII, ATP6, COIII, ND4, ND4L, Cytb and ND1), four genes start with ATT (ND2, ATP8, ND5 and ND6), ND3 gene starts with ATA, and COI starts with CGA (Table 3). Among 13 PCGs, nine genes (ND2, COI, COII, ATP8, ATP6, COIII, ND3, ND6, Cytb) are coded on the majority strand, while the rest (ND5, ND4, ND4L, ND1) are coded on the minority strand. Three PCGs (COI, COII and ND4) have incomplete stop codons consisting of a T- or TA- nucleotide, two PCGs (ND5, ND1) stop with standard terminal codon (TAT) and the other PCGs stop with standard terminal codon (TAA) (Table 4). A recent study has used expressed sequence tag to explain that COI may start with CGA [42]. COI and COII usually have an incomplete stop codon in lepidopteran species, such as in A. honmai [43], M. sexta [44], Artogeia melete [45], Phthonandria atrilineata [46], O. lunifer [47], Hyphantria cunea [48] and A. emma [49]. Between ATP8 gene and ATP6 gene of the P. c-aureum mitogenome, we found seven overlapping nucleotides which is a common feature for all lepidopteran mitogenomes known to date (Table 3).
The A+T contents of three codon positions of the PCGs were calculated and were showed in Table 5. The second position has a relatively high A +T content (80.42%), while the first and the third positions have 78.36 % and 79.02 % respectively. In addition, both the positions have negative AT-skew and postive GC-skew. Relative Synonymous Codon Usage (RSCU) for the P. c-aureum mitogenome is showed in Table 5. The results show that RSCU has a distinct bias towards T/A for 13 PCGs. Among the 64 available codons, the four most used codons are Phenylalanine (F, UUU, 11.20%), Leucine (L, UUA, 8.17%), Isoleucine (I, AUU, 8.14%), and Methionine (M, AUA, 4.77%).
Table 5: Codon usage of the protein-coding genes in Polygonia c-aureum
|
Codon(aa) |
n | % | RSCU | Codon(aa) | n | % |
RSCU |
| UUU(F) |
418 |
11.20 | 1.7 | UAU(Y) | 253 | 6.78 |
1.72 |
| UUC(F) |
74 |
1.98 | 0.3 | UAC(Y) | 41 | 1.10 |
0.28 |
| UUA(L) |
305 |
8.17 | 3.26 | UAA(*) | 248 | 6.64 |
1.55 |
| UUG(L) |
67 |
1.79 | 0.72 | UAG(*) | 72 | 1.93 |
0.45 |
| CUU(L) |
82 |
2.20 | 0.88 | CAU(H) | 50 | 1.34 |
1.72 |
| CUC(L) |
25 |
0.67 | 0.27 | CAC(H) | 8 | 0.21 |
0.28 |
| CUA(L) |
62 |
1.66 | 0.66 | CAA(Q) | 40 | 1.07 |
1.33 |
| CUG(L) |
21 |
0.56 | 0.22 | CAG(Q) | 20 | 0.54 |
0.67 |
| AUU(I) |
304 |
8.14 | 1.71 | AAU(N) | 200 | 5.36 |
1.71 |
| AUC(I) |
51 |
1.37 | 0.29 | AAC(N) | 34 | 0.91 |
0.29 |
| AUA(M) |
178 |
4.77 | 1.58 | AAA(K) | 99 | 2.65 |
1.52 |
| AUG(M) |
47 |
1.26 | 0.42 | AAG(K) | 31 | 0.83 |
0.48 |
| GUU(V) |
55 |
1.47 | 2.14 | GAU(D) | 66 | 1.77 |
1.53 |
| GUC(V) |
7 |
0.19 | 0.27 | GAC(D) | 20 | 0.54 |
0.47 |
| GUA(V) |
31 |
0.83 | 1.2 | GAA(E) | 65 | 1.74 |
1.57 |
| GUG(V) |
10 |
0.27 | 0.39 | GAG(E) | 18 | 0.48 |
0.43 |
| UCU(S) |
45 |
1.21 | 1.29 | UGU(C) | 26 | 0.70 |
1.21 |
| UCC(S) |
31 |
0.83 | 0.89 | UGC(C) | 17 | 0.46 |
0.79 |
| UCA(S) |
61 |
1.63 | 1.74 | UGA(W) | 66 | 1.77 |
1.42 |
| UCG(S) |
19 |
0.51 | 0.54 | UGG(W) | 27 | 0.72 |
0.58 |
| CCU(P) |
24 |
0.64 | 1.35 | CGU(R) | 3 | 0.08 |
0.57 |
| CCC(P) |
22 |
0.59 | 1.24 | CGC(R) | 2 | 0.05 |
0.38 |
| CCA(P) |
23 |
0.62 | 1.3 | CGA(R) | 13 | 0.35 |
2.48 |
| CCG(P) |
2 |
0.05 | 0.11 | CGG(R) | 3 | 0.08 |
0.57 |
| ACU(T) |
25 |
0.67 | 1.15 | AGU(S) | 31 | 0.83 |
0.89 |
| ACC(T) |
25 |
0.67 | 1.15 | AGC(S) | 19 | 0.51 |
0.54 |
| ACA(T) |
31 |
0.83 | 1.43 | AGA(S) | 37 | 0.99 |
1.06 |
| ACG(T) |
6 |
0.16 | 0.28 | AGG(S) | 37 | 0.99 |
1.06 |
| GCU(A) |
19 |
0.51 | 2 | GGU(G) | 20 | 0.54 |
0.82 |
| GCC(A) |
1 |
0.03 | 0.11 | GGC(G) | 4 | 0.11 |
0.16 |
| GCA(A) |
17 |
0.46 | 1.79 | GGA(G) | 53 | 1.42 |
2.16 |
| GCG(A) |
1 |
0.03 | 0.11 | GGG(G) | 21 | 0.56 |
0.86 |
A total of 3,733codons were analyzed.
RSCU, relative synonymous codon usage.
*= termination codon.
Transfer RNA and Ribosomal RNA Genes
The P. c-aureum mitogenome contains the set of 22 tRNA genes as shown in Figure 1, in which 14 tRNAs are coded on the J-strand and eight on the N-strand (Table 3). All the tRNAs have the typical clover-leaf structure, except for the tRNASer (AGN) that lacking the Dihydrouridine (DHU) arm of which forms a simple loop (Figure 3). In addition, all their anticodons are similar to those found in lepidopteran insects. We can not find a complete typical clover-leaf structure of tRNASer (AGN) by using tRNAscan-SE, as in some animal mitogenomes [50], especially in insects. The P. c-aureum mitogenome is as most of lepidopteran mitogenomes though the feature is not very conserved in the animal mitogenomes. However, there are two exceptions, showing typical clover leaf secondary structures, appeared in the tRNAs of lepidopteran insects, i.e. Diaphania pyloalis [41] and A. honmai [43]. The 22 tRNA molecules varied between 62 bp (tRNACys) and 71 bp (tRNALys) in length (Table 3), showing a highly A+T content of 80.97% and exhibiting positive AT-skew (0.006) (Table 4).
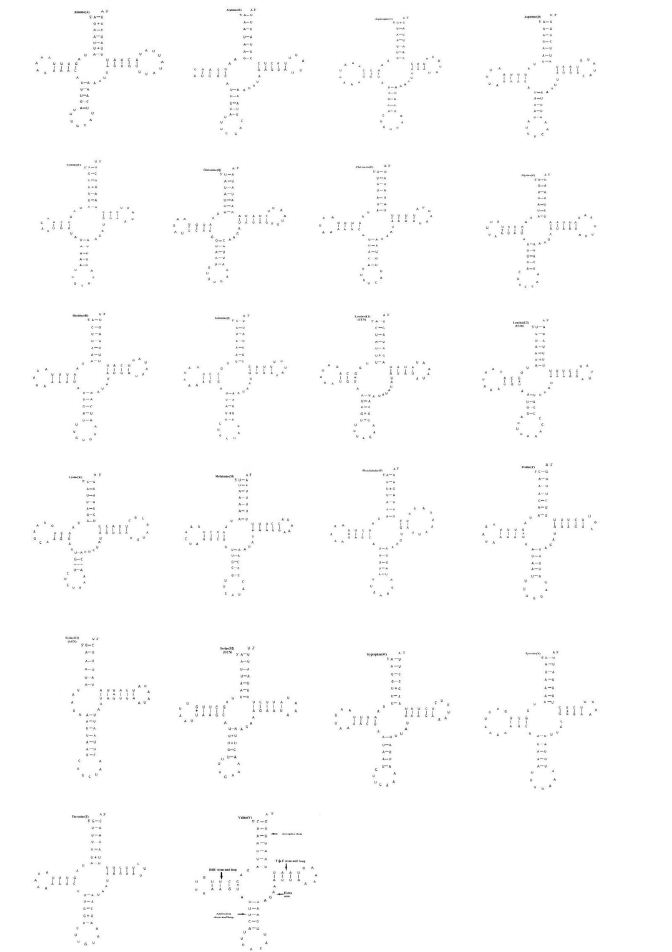
Figure 3: Predicted secondary clover-leaf structure for the 22 tRNA genes of Polygonia c-aureum. The tRNAs are labeled with the names of their corresponding amino acids. The minus sign (-) indicates Watson-Crick base pairing and the plus sign (+) indicates unmatched base pairing.
The A+T-rich Region
The A+T-rich region of P. c-aureum is 351 bp long (Table 3) with 94.02% A+T content and locates between the 16S and tRNAMet (Figure 1). This shorter region is similar to 458 bp A+T-rich region of Papilio protenor [51]. Some conserved structures found in other Nymphalidae mitogenomes were also observed in the A+T-rich region of P. c-aureum mitogenome, shown in Figure 2. It contains the motif ATAGA followed by a 19 bp poly-T stretch and contains a relatively conservative microsatellite (AT)n element (n=25). However, we did not find a poly-A (in majority strand) which is often located upstream of tRNAMet in some lepidopteran insects.
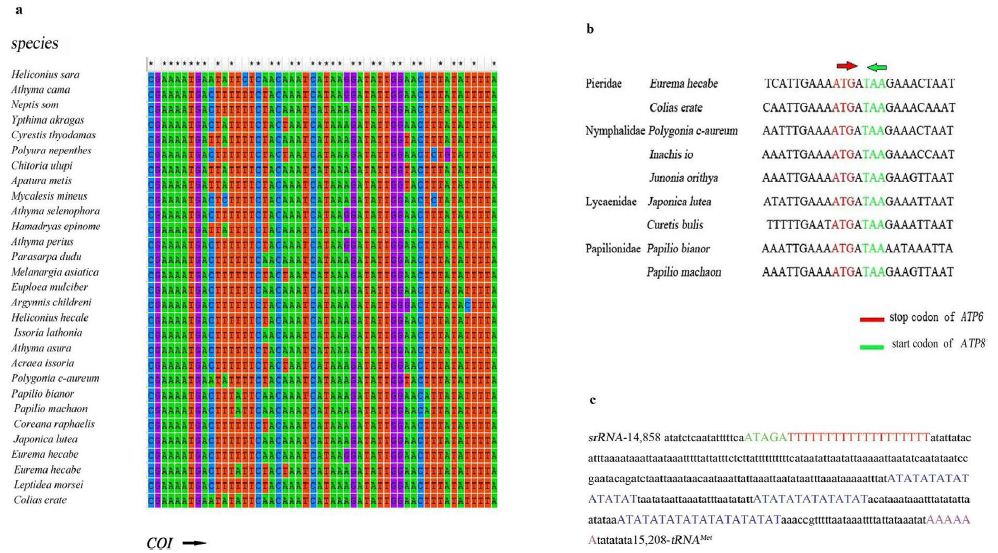
Figure 2: a) Alignment of the initiation codons of COI genes of 29 species in the study. The arrow shows the initial direction of COI genes. B) Alignment of overlapping region between ATP8 and ATP6 across Nymphalidae. C) The features present in the A+T-rich region of Polygonia c-aureum.
Phylogenetic Relationships
Different optimality criteria and dataset compilation techniques have been applied to find the best method of analyzing complex mitogenomic data [52-54]. A total of 87 available mitogenomes, including the newly sequenced mitogenome, were applied to the phylogenetic analysis (Table 1). The results of the BI and ML analyses revealed the relationships of 11 Nymphalidae subfamily lineages (Biblidinae, Apaturinae, Nymphalinae, Cyrestidinae, Limenitidinae, Heliconiinae, Satyrinae, Charaxinae, Calinaginae, Danainae and Libytheinae) with very high nodal supports, shown in Figures 4 and 5.
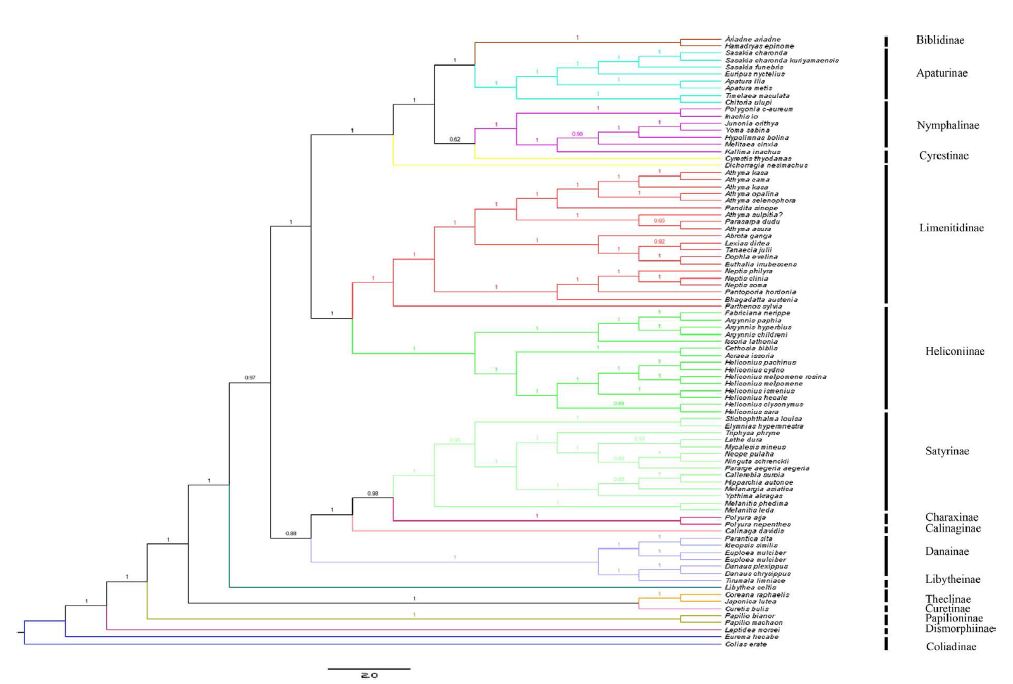
Figure 4: Phylogenetic relationship of Nymphalidae. Phylogenetic tree inferrd from nucleotide sequences of 13 PCGs using Bayesian Inference (BI) method. Number at each node show bootstrap values. The branches are coloured and their content indicated at the subfamily level.
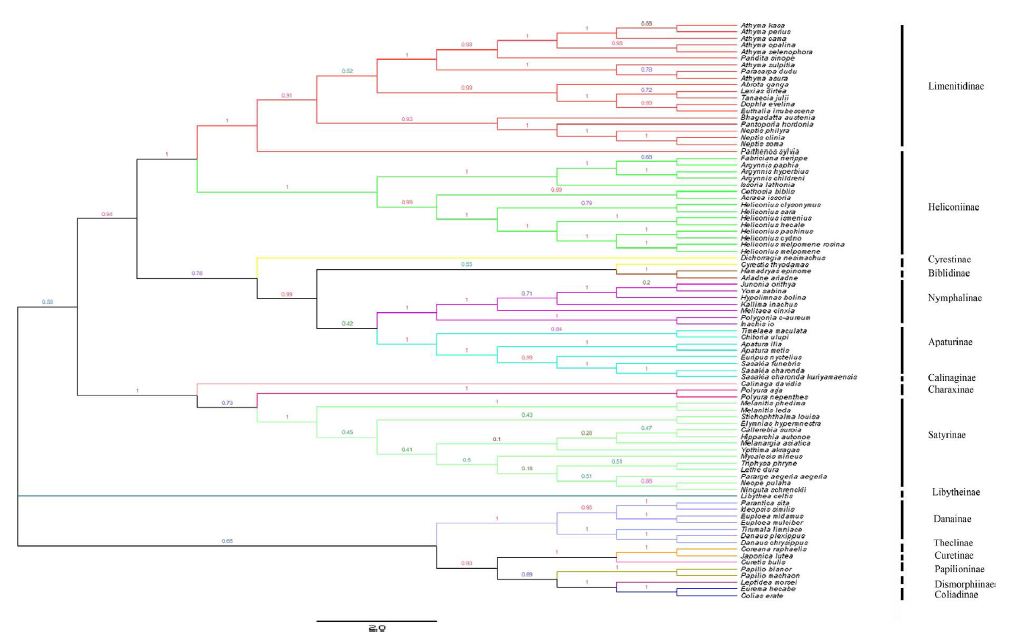
Figure 5: Inferred phylogenetic relationship among 87 species based on mitogenome sequences of 13 PCGs using Maximum Likehood (ML) method. Number at each node show bootstrap values. The branches are coloured and their content indicated at the subfamily level.
The phylogenetic analyses by BI method showed the relationships of the subfamilies of Nymphalidae, i.e. (((((Biblidinae + Apaturinae) + (Nymphalinae+ Cyrestidinae)) + (Limenitidinae + Heliconiinae)) + (((Satyrinae + Charaxinae)+ Calinaginae)+ Danainae)) + Libytheinae), with well high nodal supports. The result was consistent with the [7] whose phylogenetic analyses were based on ten nuclear genes.
Within the Nymphalidae, almost all nodes were supported by more than 0.80 supports in the BI tree. Our results showed clearly the relationships that Limenitidinae and Heliconiinae are sisters, with quite well supported by both BI (posterior probabilities =1) and ML (bootstrap =100) analyses. The results were identical to [23] and [7]. Moreover, the relationships (Calinaginae + (Charaxinae + Satyrinae)) were strongly supported by borh BI and ML trees. In addition, we found the subfamily Libytheinae located at the base of the phylogenetic tree of the Nymphalidae, which is the same as most previous hypotheses based on adult morphological studies [55-57] and molecular phylogenetic studies [7,58,59].
Though the supports were high in this study, the future studies need more samples and data to build a more powerful phylogenetic framework for Nymphalidae.
Divergence Time Estimation
The estimated divergence times among the Nymphalidae were shown in Figure 6. Our result suggested the first divergence in Nymphalidae occureded during the Cretaceous, at 89.72 Ma, and most clades appeared to have been diverged during the Cretaceous, at 86.9 Ma. The conclusion consisted with the previous result based on fossils and historical biogeography events by [38]. Besides, the Nymphalinae seems to be diverged from the group ((Biblidinae + Apaturinae) + Cyrestidinae) during the Cretaceous, at 75 Ma. These results were similar with the report of [24], and more accurated than the result of [38].
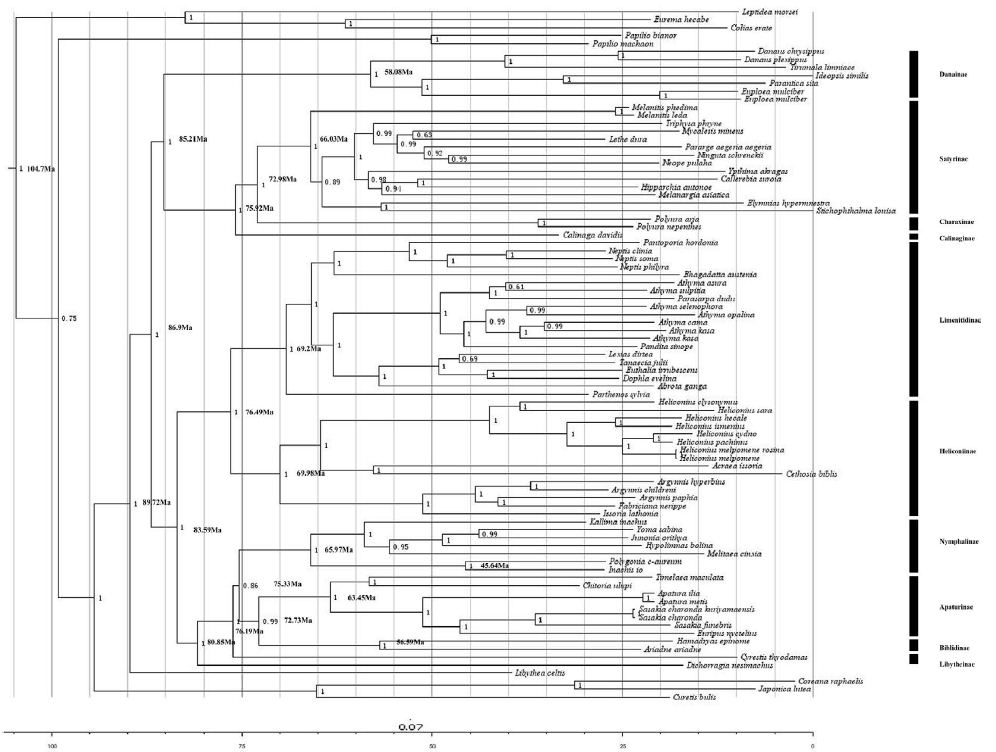
Figure 6: Estimated times of divergence for the family of Nymphalidae. The bootstrap values are shown at branching point. The time scale shows ages in million years (My) before present.
In this study, we found that the Heliconiinae clade and the Limenitidinae clade appeared to be approximately the same age about 70 Myrs. This result is consistent with the recent studies [24,40]. Our results situate the split between Limenitidinae and Heliconiinae about 69-76 Ma, which is consistent with the results of [24] who estimated this split to have occurred at 55.0-93.1 Ma. Moreover, the split between Satyrinae and Charaxinae at 66.03–72.98 Ma. We estimated that the Danainae diverged from the group (Calinaginae+ (Charaxinae + Satyrinae)) to be situated between 85–75 Ma, consistent with [24]. The Libytheinae arised as basal to the Nymphalidae diverged from the other subfamilies of Nymphalidae at 87.92 Ma. This is also consistent with [24]. In addition,for the first time, our analyses suggest that the genus Polygonia began to diversify, with the other lineage off from the common ancestor of the rest of Nymphalinae, at about 45.64 Ma.
Conclusions
In summary, we have shown that a complete mitogenome of the Asian comma butterfly, P c-aureum. The formerly identified conserved elements of Lepidoptera mitogenomes, i.e. the motif ‘ATAGA’ and poly-T stretch in the A+T-rich region, the long intergenic spacer upstream of ND2 and the 7 bp overlapping between ATP8 and ATP6, are present in P. c-aureum, only with some subtle differences in both of the size of genes and of the intergenic regions. The phylogenetic relationships based on nucleotide sequences of 13 PCGs by using BI and ML methods clarified the taxonomic status of Nymphalidae with a robust support. Furthermore, our results indicated that the complete mitogenome can be as an effective molecular marker to resolve the relationships of subfamilies within a family of butterflies. Our research is consistent with previous studies on the phylogenetic relationships of Nymphalidae. For the first time, we found that the genus Polygonia began to diversify at about 45.64 Ma. In addition, as in previous molecular studies, the subfamilies within Nymphalidae maybe diverged from each other in the Early Cretaceous, at about 90 Ma. We hope our results would be useful for the further phylogenetic analyses of insects and for the prevention and control of insect pests as well. Consequently, excellent phylogenetic resolution will come from larger integrated datasets. Predicatively, greater integration of nuclear and mitogenome studies is necessary to further our understanding for insect evolution.
Acknowledgments
This work was supported by grants from the Natural Science Foundation of China (No. 31572246) to Guo-Fang Jiang.
References
- Chou I (1994) Monograph of Chinese Butterflies (in Chinese). Henan Scientific and Technological Publishing House.
- Chou I (1998) Classification and Identification of Chinese Butterflies (in Chinese). Henan Scientific and Technological Publishing House.
- Freitas AVL, Oliveira PS (1992) Biology and behavior of the neotropical butterfly Eunica bechina (Nymphalidae) with special reference to larval defence against ant predation. Journal of Research on the Lepidoptera 31: 1-11.
- Braby MF (1994) Phenotypic variation in adult Mycalesis Hübner (Lepidoptera: Nymphalidae: Satyrinae) from the Australian wet-dry tropics. Australian Journal of Entomology 33: 327-336.
- Wahlberg N, Weingartner E, Nylin S (2003) Towards a better of the understanding higher systematics of Nymphalidae (Lepidoptera: Papilionoidea). Molecular Phylogenetics & Evolution 28: 473-484. [crossref]
- Freitas AVL, Brown KS (2004) Phylogeny of the Nymphalidae (Lepidoptera). Systematic Biology 53: 363-383. [crossref]
- Wahlberg N, Leneveu J, Kodandaramaiah U, Peña C, Nylin S, et al. (2009) Nymphalid butterflies diversify following near demise at the Cretaceous/Tertiary boundary. Proceedings Biological Sciences 276: 4295-4302.
- Brower AVZ, Wahlberg N, Ogawa JR, Boppré M, Vane-Wright RI (2010) Phylogenetic relationships among genera of danaine butterflies (Lepidoptera: Nymphalidae) as implied by morphology and DNA sequences. Systematics and Biodiversity 8: 75-89.
- Penz CM, Mohammadi N, Wahlberg N (2011) Neotropical Blepolenis butterflies: wing pattern elements, phylogeny, and Pleistocene diversification (Lepidoptera, Nymphalidae). Zootaxa 17: 5326.
- Ehrlich PR, Raven PH (1964) Butterflies and plants: a study in coevolution. Evolution 18: 586-608.
- Hanski I (1999) Metapopulation ecology. Oxford University Press.
- Beldade P, Koops K, Brakefield PM (2002) Developmental constraints versus flexibility in morphological evolution. Nature 416: 844-847.
- Brower AVZ (1996) A new mimetic species of Heliconius (Lepidoptera: Nymphalidae), from southeastern Colombia, revealed by cladistic analysis of mitochondrial DNA sequences. Zoological Journal of the Linnean Society 116: 317-332.
- Mallet J, McMillan WO, Jiggins CD (1998) Mimicry and warning color at the boundary between races and species. In: Endless forms: species and speciation. New York: Oxford 390-403.
- Forbes WTM (1928) Variation in Junonia lavinia (Lepidoptera: Nymphalidae). Journal of the New York Entomological Society 36: 305-320.
- Silberglied RE (1984) Visual communication and sexual selection among butterflies. Symposia of the Royal Entomological Society of London.
- Dasmahaptra KK, Blum MJ, Aiello A, Hackwell S, Davies N, et al. (2002) Inferences from a rapidly moving hybrid zone. Evolution 56: 741-753. [crossref]
- Austin GT, Murphy DD, Baughman JF, Launer AE, Fleishman E (2003) Hybridization of checkerspot butterflies in the Great Basin. Journal of the Lepidopterists Society 57: 176-192.
- Li CL, Zhu BY (1992) The profile of butterflies in China (in Chinese). Shanghai Yuandong Press.
- Wahlberg N, Brower AVZ, Nylin S (2005) Phylogenetic relationships and historical biogeography of tribes & genera in the subfamily Nymphalinae (Lepidoptera: Nymphalidae). Biological Journal of the Linnean Society 86: 227-251.
- Ackery, P.R. (1988) Hostplants and classification: a review of nymphalid butterflies. Biological Journal of the Linnean Society 33: 95-203.
- Kuznetzov VI, Stekolnikov AA (2001) New approaches to the system of Lepidoptera of world fauna. Proceedings of the Zoological Institute of St. Petersburg 282: 1-462.
- Brower AVZ (2000) Phylogenetic relationships among the Nymphalidae (Lepidoptera) inferred from partial sequences of the wingless gene. Proceedings Biological Sciences 267: 1201-1211. [crossref]
- Pohl N, Sison-Mangus MP, Yee EN, Liswi SW, Briscoe AD (2009) Impact of duplicate gene copies on phylogenetic analysis and divergence time estimates in butterflies. BMC Evolutionary Biology 9: 99-115. [crossref]
- Shen RW, Wang JG, Zhan GX (1991) Studies on the biology of Polygoniac-aureum Acta Agriculturae Universitatis Jiangxiensis 1: 3.
- Simon C, Frati F, Bekenbach A, Crespi B, Liu H, et al. (1994) Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chainreaction primers. Annals of the Entomological Society of America 87: 651-701.
- Simon C, Buckley TR, Frati F, Stewart JB, Beckenbach AT (2006) Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annual Review of Ecology, Evolution & Systematics 37: 545-579.
- Singh, V.K., Mangalam, A.K., Dwivedi, S. & Naik, S. (1998) Primer premier: program for design of degenerate primers from a protein sequence. Biotechniques, 24, 318-319. [crossref]
- Burland TG (1999) DNASTAR’s lasergene sequence analysis software. Bioinformatics Methods and Protocols 132: 71-91.
- Kumar S, Stecher G, Tamura K (2016) MEGA7: Molecular evolutionary genetics analysis version7.0 for bigger datasets. Molecular Biology and Evolution 33: 1870-1874. [crossref]
- Junqueira AC, Lessinger AC, Torres TT, Silva FR, Vettore AL, et al. (2004) The mitochondrial genome of the blowfly ChrysoMa chloropyga (Diptera: Calliphoridae). Gene 339: 7-15. [crossref]
- Lowe TM, Chan PP (2016) tRNAscan-SE On-line: integrating search & context for analysis of transfer RNA genes.Nucleic Acids Research 44: W54-57. [crossref]
- Wickware P (1997) DNasis for Windows—Sequence Analysis Software User’s Manual. Hitachi Software Genetic Systems 25-211.
- Steinberg S, Cedergren R (1994) Structural compensation in atypical mitochondrial tRNAs. Nature Structural Biology 1: 507-510. [crossref]
- Nylander JAA (2004) MrModeltest v2. Program distributed by the author. Evolutionary Biology Centre.
- Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19: 1572-1574. [crossref]
- Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu CH, et al. (2014) BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Computational Biology 10: e1003537. [crossref]
- Wahlberg N (2006) That awkward age for butterflies: insights from the age of the butterfly subfamily Nymphalinae (Lepidoptera: Nymphalidae). Systematic Biology 55: 703-714. [crossref]
- Peñalver, E. & Grimaldi, D.A. (2006) New data on Miocene butterflies in Dominican amber (Lepidoptera: Riodinidae and Nymphalidae) with the description of a new nymphalid. American Museum Novitates 3519: 1-17.
- Wahlberg N, Wheat CW, Peña C (2013) Timing and patterns in the taxonomic diversification of Lepidoptera (butterflies and moths). PLoS ONE 8: e80875.
- Zhu BJ, Liu QN, Dai LS, Wang L, Sun Y, et al. (2013) Characterization of the complete mitochondrial genome of Diaphania pyloalis (Lepidoptera: Pyralididae). Gene 527: 283-291. [crossref]
- Margam VM, Coates BS, Hellmich RL, Agunbiade T, Seufferheld MJ (2011) Mitochondrial genome sequence and expression profiling for the legume pod borer Maruca vitrata (Lepidoptera: Crambidae). PLoS ONE 6: e16444. [crossref]
- Lee ES, Shin KS, Kim MS, Park H, Cho S, et al. (2006) The mitochondrial genome of the smaller tea tortrix Adoxophyes honmai (Lepidoptera: Tortricidae). Gene 373: 52-57. [crossref]
- Cameron SL, Whiting MF (2008) The complete mitochondrial genome of the tobacco hornworm, Manduca sexta (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene 408: 112-123. [crossref]
- Hong G, Jiang S, Yu M, Yang Y, Li F, et al. (2009) The complete nucleotide sequence of the mitochondrial genome of the cabbage butterfly, Artogeia melete (Lepidoptera: Pieridae). Acta Biochimical et Biophysical Sinica 41: 446-455.
- Yang L, Wei ZJ, Hong GY, Jiang ST, Wen LP (2009) The complete nucleotide sequence of the mitochondrial genome of Phthonandria atrilineata (Lepidoptera: Geometridae). Molecular Biology Reports 36: 1441-1449. [crossref]
- Salvato P, Simonato M, Battisti A, Negrisolo E (2008) The complete mitochondrial genome of the bag-shelter moth Ochrogaster lunifer (Lepidoptera, Notodontidae). BMC Genomics 9: 331. [crossref]
- Liao F, Wang L (2010) The complete mitochondrial genome of the fall webworm, Hyphantria cunea (Lepidoptera: Arctiidae). International Journal of Biological Sciences 6: 172.
- Lu HF, Su TJ, Luo AR, Zhu CD, Wu CS (2013) Characterization of the complete mitochondrion genome of diurnal moth Amata emma (Butler) (Lepidoptera: Erebidae) and its phylogenetic implications. PLoS ONE 8: e72410. [crossref]
- Wolstenholme DR (1992) Animal mitochondrial DNA: structure and evolution. International Review of Cytology 141: 173-216. [crossref]
- Liu NY, Wu YH, Yang XJ, Wu J, Zheng SZ, et al. (2017) Complete mitochondrial genome of Papilio protenor (Lepidoptera, Papilionidae) & implications for Papilionidae taxonomy. Journal of Insect Science 17: 1-8.
- Castro LR, Dowton M (2005) The position of the Hymenoptera within the Holometabola as inferred from the mitochondrial genome of Perga condei (Hymenoptera: Symphyta: Pergidae). Molecular Phylogenetics and Evolution 34: 469-479. [crossref]
- Kim I, Lee EM, Seol KY, Yun EY, Lee YB, et al. (2006) The mitochondrial genome of the Korean hairstreak, Coreana raphaelis (Lepidoptera: Lycaenidae). Insect Molecular Biology 15: 217-225. [crossref]
- Stewart JB, Beckenbach AT (2005) Insect mitochondrial genomics: the complete mitochondrial genome sequence of the meadow spittlebug Philaenus spumarius (Hemiptera: Auchenorrhyncha: Cercopoidae). Genome 48: 46-54. [crossref]
- Ehrlich PR (1958) The morphology, phylogeny, and higher classification of butterflies (Lepidoptera: Papilionoidea). University of Kansas Science Bulletin 39: 305-370.
- Scott, J.A. (1984) The phylogeny of butterflies (Papilionoidea and Hesperoidea). Journal of Research on the Lepidoptera 23: 241-281.
- De Jong R, Vane-Wright RI, Ackery PR (1996) The higher classification of butterflies (Lepidoptera): problems and prospects. Insect Systematics Evolution 27: 65-101.
- Weller SJ, Pashley DP, Martin JA (1996) Reassessment of butterfly family relationships using independent genes and morphology. Annals of the Entomological Society of America 89: 184-192.
- Wahlberg N, Klemetti T, Selonen V, Hanski I (2002) Metapopulation structure and movements in five species of checkerspot butterflies. Oecologia 130: 130-343.