DOI: 10.31038/CST.2017223
Abstract
Background: HDAC inhibition is known to modulate expression of tumour suppressor genes and induce cell differentiation, growth arrest and apoptosis. The aim of this study was to evaluate the efficacy of a novel series of hydroxamic acid based HDAC inhibitors in cell based assays and tumour xenograft models.
Material and Methods: A series of novel hydroxamate derivatives were synthesized and evaluated. HDAC enzyme inhibitory activity was measured using Hela nuclear extracts. Anti-proliferative activity was assessed in a panel of cancer cell lines. Anti-apoptotic activity was evaluated by caspase-3 activation. In vivo efficacy was evaluated in lung adenocarcinoma xenograft model.
Results: The compounds showed potent HDAC inhibitory activity and anti-proliferative activity in several cancer cell lines. In an in vivo A549 lung xenograft model, the compounds exhibited significant tumor growth inhibition.
Conclusion: The novel HDAC inhibitors showed anti-proliferative activity against several human cancer cell lines and also anti-tumor activity in a Mouse xenograft model.
Keywords:
Histone deacetylation, HDAC inhibitor, Vorinostat
Introduction
Histone acetylation/deacetylation is mediated by a class of enzymes known as Histone acetyl transferase (HATs) and histone deactylases (HDACs). HDACs (histone deacetylases) are important enzymes in the regulation of gene expression in eukaryotic cells [1]. HDACs are key enzymes involved in the regulation of histone and non-histone proteins [2]. Increased levels of HDACs in tumor cells are known to be closely associated with tumor initiation, progression and metastasis [3-4]. Inhibition of Histone deacetylases is known to play an important role in epigenetic regulation by inducing cell death, apoptosis, and cell cycle arrest in cancer cells.
Histone deacetylase (HDAC) inhibitors are a diverse group of small molecule drugs that induce a broad range of effects on cancer cells, including cell cycle arrest, apoptosis, cell differentiation, autophagy and anti-angiogenic effects [5]. Histone deacetylase inhibitors (HDACi) have emerged as a class of therapeutic agents that induce tumor cell cytostasis, differentiation and apoptosis in various hematologic and solid malignancies [6-7]. These drugs inhibit HDAC, and several of them have been developed as anti-cancer agents as they have a significant effect specifically on tumor-cell proliferation compared to non-malignant cells.
HDACs inhibitors can be divided into four major structural classes: (1) small molecular weight carboxylates; (2) hydroxamic acids; (3) benzamides; and (4) cyclic peptides [8-9]. Vorinostat (Zolinza) and Romidepsin (Istodax) are the only HDACs inhibitors currently approved by the U.S. Food and Drug Administration (FDA) for the treatment of refractory cutaneous T-cell lymphoma (CTCL) [10-11].
Most of the HDAC inhibitors that have entered clinical trials have limitations, including low bioavailability, low potency, cardiovascular safety issues, and potential for drug-drug interactions through cytochrome P450 inhibition [12]. Therefore, there is still a clinical opportunity for novel, orally available efficacious HDAC inhibitors with a wider safety margin. HDAC inhibitors exhibit anti-proliferative activity in both in vitro and in vivo pre-clinical models of cancer, with many of them being evaluated as anticancer therapeutics in the clinic. However, keeping in mind the poor bioavailability and efficacy in solid tumors, there still remains an unmet medical need to discover newer HDAC inhibitors derived from novel structural classes of compounds.
In the present paper, we report the identification of novel hydroxamic acid based small-molecule HDAC inhibitors by mediumthroughput screening of a compound library using a fluorescence based assay with Hela Nuclear extracts as the enzyme source. A focused library of about 100 compounds was designed and synthesized, among which several compounds showed equivalent or higher potencies against HDAC as compared to Vorinostat. The hit compounds from the primary screening were evaluated for their effects on cellular proliferation in a panel of human cancer cell lines. We identified four compounds which showed a potent GI50 in the low micromolar range. Several of these novel HDAC inhibitors could be promising new lead structures for further development as improved anticancer drugs. In conclusion, the screening of a library of compounds for HDAC inhibitory activity and anti-proliferative effect in cancer cells has identified several promising new leads for further development.
Materials and methods:
Synthesis of HDAC inhibitors
The compound library was obtained from the Medicinal chemistry department at Anthem Biosciences.
Based on general formula 1, about 100 hydroxamic acid derivatives were designed and synthesized.
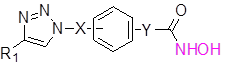
General formula I
These compounds were synthesized as described in below synthetic scheme 1.
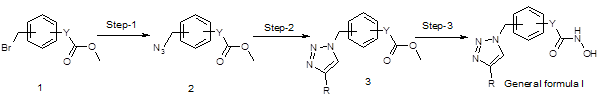
Scheme 1
Reactions of the compound 1 with sodium azide in dimethylformamide at 40 oC resulted compound 2. Then the compound 3 was synthesized by standard click chemistry by reacting the compound 2 with appropriate alkyne in the presence of copper iodide and Hunig’s base in dimethylformamide [13]. Reaction of the compound 3 with hydroxyl amine in the presence of suitable bases such as sodium methoxide in methanol yielded the compounds of general formula I.
Out of nearly 100 compounds synthesized, 4 compounds i.e. PAT-1101, PAT-1103, PAT-1118 and PAT-1125 were identified as hit molecules from the primary screening. Suitable salts of the four compounds were prepared to improve their drug like properties.
Preparation of Hela Nuclear extracts:
Nuclear fractions prepared from Hela cells as per established protocols were used as a source of HDAC enzyme. Hela cells obtained from ATCC were cultured in complete growth medium containing 10% fetal bovine serum supplemented with antibiotics. Subconfluent cells were harvested and washed in phosphate buffered saline (PBS). 1×107 cells were resuspended in 1 mL of cold lysis buffer containing 10 mM Tris HCl (pH 7.5), 10 mM NaCl, 15 mM MgCl2, 250 mM Sucrose, 0.1 mM EGTA and 0.5% NP-40. The cell lysate was then maintained on ice for 15 min. To the lysate, 4 mL of Sucrose buffer containing 30% sucrose, 10 mM Tris HCl (pH 7.5), 10 mM NaCl and 3 mM MgCl2 was added and the resultant mixture was centrifuged at 1500 rpm for 10 min at 4 deg C. The resultant pellet was resuspended in 1 mL of Tris-HCl buffer (10 mM Tris-HCl, pH 7.5, 10 mM NaCl) and recentrifuged at 1500 rpm for 10 min at 4 deg C. The resultant supernatant was discarded and the isolated nuclei was resuspended in 100 µL of cold extraction buffer containing 50 mM HEPES pH 7.5, 420 mM NaCl, 5 mM EDTA, 1mM EGTA, and 10% glycerol. The solution was sonicated for 30 sec and incubated on ice for 30 min following which it was centrifuged at 10000 rpm for 10 min at 4 deg C. The supernatant was collected and used as the enzyme source for HDAC assay.
In vitro HDAC inhibition Assay:
HDAC inhibition assay was performed using a fluorescence based assay with a fluorescent substrate (Boc-Lys (Ac)-AMC Substrate) as reported previously [14-15]. Stock solutions of the compounds were prepared in 100% DMSO. 3 µg of the nuclear extracts was preincubated with the compounds for 10 min at 30 deg C. The substrate was diluted in 50 µL of assay buffer (25 mM Tris HCl pH 8.0, 137 mM NaCl, 2.7 mM KCl, 1mM MgCl2) and added to a 96-well plate. The plate was incubated for a further 45 min at 30 deg C. The reaction was terminated by the addition of 50 µL of developer and incubated for 15 min at 30 deg C. The fluorescent deacetylated substrate was detected at lexc of 340/40 and lemi of 460/40 using a Microplate Reader (BioTek Instrument Inc.). The fluorescent signal was compared with the DMSO treated wells and the percentage inhibition was determined. IC50 (50% HDAC inhibitory concentration) was determined by testing in a wide concentration range of 0.001, 0.01, 0.1, 1 and 10μM.
Cell proliferation assays:
Anti-proliferative activity of the compounds was tested against a panel of cancer cell lines including Lung, Cervix, Colon, Brain, Renal, Leukemia, Prostate, Pancreas, Skin, Bone, Breast, Ovary cancer by using a standard MTT assay. Human cancer cell lines (American Type Culture Collection) were cultured in complete media containing 10% heat inactivated fetal bovine serum and 100 U/ml Penicillin, 100 µg/ml Streptomycin in a 37°C, 5% CO2 humidified incubator and passaged twice weekly. Cells were seeded in 96-well plates at a density of 3X103 cells per well in 100 µL and were allowed to attach for 24 h. Stock concentrations of the compounds were made in DMSO. 100 µL of media containing various concentrations of compounds (1, 10 and 100 µM) were added to the cells and were incubated for 48 hours. Vorinostat was tested as a reference compound in the assay. On the day of termination, 50 µL of MTT (3-(4,5-dimethylthiazol- 2-yl)-2,5-diphenyl-2H-tetrazolium bromide) (Sigma, St Louis, MO, USA) solution (5mg/mL) was added to the medium and the cells were incubated for 3 hours. The medium was then aspirated and 100% DMSO was added to solubilize the violet MTT-formazan product. The absorbance at 570 nm was measured on a 96-well plate reader by spectrophotometry (Biotek Synergy HT). Assays were performed in duplicates for each concentration. Results are expressed as percentage of growth inhibition with respect to the DMSO treated control wells. A dose response curve was generated and GI50 values were interpolated from the growth curves using GraphPad Prism software.
Apoptosis assay (Caspase-3 activation):
Caspase-3 activity was measured in HT-29 cells using a commercially available kit (Sigma- Aldrich). Briefly, HT-29 cells were cultured in McCoy’s 5a medium containing 10% FCS and antibiotics. On the day of the study 10,000 cells were seeded into each well of a 96 well plate and incubated for 12-16 h. The compounds were added at concentrations ranging from 0.1 µM to 30 µM and incubated for 48 h. The cells were then lysed in lysis buffer and the lysates were used to perform the assay according to the manufacturer’s instruction. The assay is based on the hydrolysis of acetyl Asp-Glu-Val-Asp 7-amido- 4-methylcoumarin (Ac-DEVD-AMC) by caspase 3, resulting in the release of the fluorescent 7-amino-4-methylcoumarin (AMC) which is measured at an excitation and emission wavelength of 360 nm and 460 nm respectively.
In vivo anti-tumor activity in A549 lung adenocarcinoma xenograft model:
All experimental procedures involving animals were approved by the Institutional Animal Ethics Committee of Anthem Biosciences. In vivo anti-tumor activity of the HDAC inhibitors was assessed in 6 week old Athymic Nude mice. Animals were purchased from Harlan Laboratories, Indianapolis IN (presently Envigo) and housed in individually ventilated cages under controlled conditions and maintained on a 12-h light/12-h dark cycle, with food and water supplied ad libitum. A549 (lung adenocarcinoma) obtained from ATCC were cultured in RPMI-1640 growth medium containing 10% FBS and antibiotics. Sub-confluent monolayers were harvested and a cell suspension of >90% viability was prepared in 1X HBSS, pH 7.4 (Hank’s Balanced Salts Solution, Sigma) and mixed with an equal volume (1: 1) of ice cold Matrigel® (Corning Life Sciences). 0.1 mL of the cell suspension containing 1×106 cells was injected into the flank region of the animals under isoflurane anesthesia. Animals were monitored daily during the period between inoculation and palpable tumor growth. Tumor volume was calculated using the formula, Tumor volume = (length × width2)/2. Tumor bearing mice were randomized into control and treatment groups (n=8) when the tumor volume reached ~100 mm3. The compounds were formulated in a vehicle containing 0.5% Carboxy methyl cellulose and 0.1% Tween 80 in water and administered by oral gavage to tumor bearing mice once daily for 21 days. The compounds were tested at 150 mg/kg. The Control group received the vehicle alone. Clinical signs were observed daily and tumor volume and was body weight was measured twice weekly during the study.
Data Analysis:
The terminal tumor volumes from in vivo xenograft studies were subjected to one-way ANOVA analysis followed by Dunnett’s test when there were multiple treatment groups. Results were considered statistically significant when P < 0.05.
Results
HDAC enzyme inhibition:
The biological activity of the HDAC inhibitors was assessed in vitro using a cell free HDAC enzymatic assay. Several compounds exhibited potent HDAC-inhibitory activity with IC50 values of in the nanomolar range (Table 1). Our results indicate that the in vitro HDAC inhibition potency is higher than the reference compound Vorinostat (Figure 1).
Table I. Characterization of hit compounds
| Compound | M. Wt. (g/mol) | Molecular
Formula |
HDAC IC50 (nM) |
| PAT-1101 | 403.49 | C23H25N5O2.HCl | 4 |
| PAT-1103 | 419.49 | C23H25N5O3.HCl | 1 |
| PAT-1118 | 393.45 | C21H23N5O3.HCl | 23 |
| PAT-1125 | 377.45 | C21H23N5O2.HCl | 4 |
| Vorinostat | 264.32 | C14 H20 N2 O3 | 78 |
HDAC inhibitory activity of synthesized compounds was measured using Hela cell nuclear extract as the enzyme source by a fluorescence based assay as described under the Materials and Methods section. IC50 was calculated from concentration versus percentage inhibition plotted using Graph Pad Prism software.
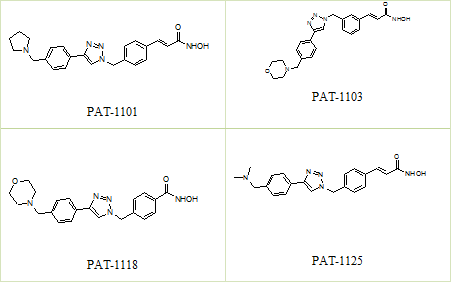
Figure 1. Structures of compounds
Anti-proliferative activity in cancer cells:
The growth-inhibitory activity of 4 compounds identified through the primary HDAC inhibitory screening was assessed in vitro in a panel of Human cancer cell lines. Cells were treated with the HDAC inhibitors at various concentrations and GI50 was determined. All the 4 compounds identified resulted in a dose-dependent inhibition of cellular proliferation at low micro molar concentrations in most of the cell lines tested (Table 2). The inhibitory effect of PAT-1101, PAT- 1103, PAT-1118, PAT-1125 on the proliferation of cancer cells was comparable or superior to that of Vorinostat against several cell lines under our experimental conditions.
Table 2. Anti-proliferative activity of hit compounds expressed as Mean Growth inhibitory concentration (GI50 in µM) in a panel of cancer cell lines
| Tissue | Cell line | Growth inhibition: GI50 concentration (µM) | ||||
| PAT-1101 | PAT-1103 | PAT-1118 | PAT-1125 | Vorinostat | ||
| Colon
|
Colo-205 | 0.31 ± 0.06 | 0.2 ± 0.03 | 0.3 ± 0.071 | 0.4 ± 0.04 | 1.6 ± 0.1 |
| HCT-116 | 0.10 ± 0.11 | 0.2 ± 0.05 | 1.2 ± 0.018 | 0.2 ± 0.04 | 2.2 ± 0.0 | |
| HT-29 | 0.15 ± 0.13 | 0.4 ± 0.13 | 1.5 ± 0.65 | 2.1 ± 0.46 | 3.6 ± 2.6 | |
| Lung | A549 | 1.58 ± 1.1 | 2.0 ± 1.52 | 4.5 ± 2.33 | 2.2 ± 2.22 | 5.9 ± 2.4 |
| NCI-H23 | 0.52 ± 0.04 | 0.4 ± 0.09 | 2.3 ± 1.14 | 0.5 ± 0.24 | 3.2 ± 1.0 | |
| NCI-H460 | 0.3 ± 0.04 | 0.45 ± 0.28 | 2.4 ± 0.85 | 1.9 ± 0.74 | 4.4 ± 1.4 | |
| Prostate | DU-145 | 0.079 ± 0.04 | 0.1 ± 0.03 | 0.3 ± 0.124 | 0.1 ± 0.04 | 1.3 ± 0.0 |
| PC-3 | 3.5 ± 2.7 | 1.7 ± 1.02 | 13.4 ± 3.3 | 4.8 ± 1.4 | 8.3 ± 0.6 | |
| Ovary | SK-OV-3 | 0.04 ± 0.016 | 0.5 ± 1.2 | 1.8 ± 0.20 | 1.7 ± 0.16 | 4.4 ± 1.5 |
| PA-1 | 0.08 ± 0.04 | 0.04 ± 0.01 | 0.2 ± 0.037 | 0.1 ± 0.01 | 0.3 ± 0.05 | |
| Cervix | Ca Ski | 0.42 ± 0.15 | 0.4 ± 0.28 | 1.2 ± 0.22 | 1.1 ± 0.73 | 6.0 ± 5.3 |
| Hela-229 | 1.0 ± 0.27 | 0.6 ± 0.38 | 1.4 ± 0.26 | 0.7 ± 0.47 | 4.7 ± 3.2 | |
| Hela-S3 | 0.23 ± 0.12 | 0.2 ± 0.02 | 0.6 ± 0.16 | 0.2 ± 0.08 | 2.9 ± 0.5 | |
| Brain | IMR-32 | 0.25 ± 0.07 | 0.2 ± 0.02 | 0.9 ± 0.201 | 0.2 ± 0.04 | 1.8 ± 0.2 |
| U-87-MG | 0.76 ± 0.66 | 1.1 ± 0.73 | 2.7 ± 0.74 | 1.0 ± 0.33 | 6.7 ± 1.1 | |
| SH-SY-5Y | 0.34 ± 0.033 | 0.2 ± 0.03 | 0.2 ± 0.06 | 0.1 ± 0.0 | 0.8 ± 0.4 | |
| Breast | MCF-7 | 3.5 ± 2.6 | 2.1 ± 2.63 | 5.7 ± 1.62 | 5.5 ± 1.2 | 5.5 ± 1.2 |
| Renal | ACHN | 0.09 ± 0.02 | 0.2 ± 0.08 | 0.7 ± 0.11 | 0.1 ± 0.04 | 1.6 ± 0.5 |
| 786-O | 2.65 ± 0.08 | 1.5 ± 0.70 | 2.5 ± 1.01 | 2.0 ± 1.1 | 4.4 ± 2.0 | |
| Leukemia | RPMI-8226 | 0.15 ± 0.05 | 0.2 ± 0.25 | 0.5 ± 0.43 | 0.3 ± 0.16 | 2.2 ± 2.3 |
| K562 | 0.19 ± 0.05 | 0.2 ± 0.08 | 0.3 ± 0.074 | 0.2 ± 0.04 | 2.2 ± 0.7 | |
| Pancreas | PANC-1 | 1.03 | 4 | 6.6 | 1.4 | 21.9 |
| Skin | A431 | 0.28 ± 0.05 | 0.2 ± 0.03 | 1.2 ± 0.533 | 0.4 ± 0.08 | 1.9 ± 1.0 |
| Bone | KHOS | 11.7 ± 1.1 | 4.6 ± 0.55 | 2.8 ± 0.36 | 10.3 ± 3.26 | 29.3 ± 7.2 |
Anti-proliferative effect of the compounds in a panel of cancer cell lines obtained from ATCC using MTT reagent as described under Materials and Methods. Results are represented as GI50 or the concentration of the compound which inhibits 50% of cell growth. GI50 was calculated using GraphPad Prism software. The Mean GI50 was derived from individual assays performed in triplicates
Induction of Apoptosis in Human Cancer Cells:
We further investigated the compounds’ ability to induce apoptosis in cancer cells and to determine if the apoptosis was caspase dependent. In this regard, compound induced Caspase-3 activation in HT-29 cells was measured using a fluorescence based assay. The compounds significantly activated caspase-3 enzyme with an EC50 which was similar to that of Vorinostat (Table 3).
Table 3.Caspase-3 activation assay
| Compound | EC50 (µM) |
| PAT-1101 | 6.59 |
| PAT-1103 | 3.62 |
| PAT-1118 | 1.50 |
| PAT-1125 | 4.09 |
| Vorinostat | 4.52 |
Anti-tumor Activity in Human Tumor Xenograft Models:
To assess the tumor growth inhibitory activity of the HDAC inhibitors in vivo, we evaluated their effect in a subcutaneous human xenograft model in Athymic Nude mice. The anti-tumor efficacy was evaluated in mice engrafted with A549 lung adenocarcinoma cells. Once daily oral administration of PAT-1103, PAT-1118 and PAT-1125 resulted in a significant tumor growth inhibition (TGI) after 21 days (Figure 2). The tumor growth inhibition (TGI) achieved as a result of treatment was between 67 and 69% at 150 mg/Kg dose. The efficacy was superior to that of Vorinostat which produced 54% tumor growth inhibition at the same dose (Table 4). Furthermore, there were no adverse clinical signs and no significant reduction in body weight in the treated mice compared to the vehicle control group.
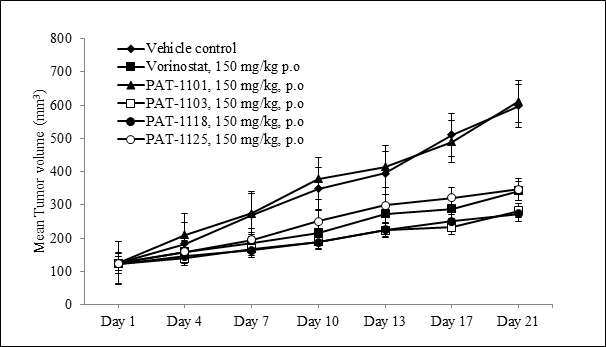
Figure 2. Anti-tumor efficacy of HDAC inhibitors in A549 lung tumor xenograft
Tumor growth kinetics in Nude mice subcutaneously implanted with 1×106 A549 cells and treated with HDAC inhibitors or Vorinostat at 150 mg/kg, p.o,( n=8 in each group) once daily for 21 days.
Table 4.Tumor growth Inhibition (TGI) in subcutaneous A549 lung tumor xenograft models established in Nude mice
| Compound | Dose (mg/Kg, p.o) | Mean Tumor volume (mm3) on Day 21 | % TGI |
| Control (Vehicle) | 0 | 598.5 ± 72.9 | – |
| PAT-1101 | 150 | 611.8 ± 125.4 | 3 |
| PAT-1103 | 150 | 282.0 ± 34.7** | 68 |
| PAT-1118 | 150 | 272.4 ± 42.8** | 67 |
| PAT-1125 | 150 | 346.4 ± 67.9** | 69 |
| Vorinostat | 150 | 342.10 ± 42.80* | 54 |
TGI: Tumor growth inhibition was calculated with respect to the Vehicle treated Control group on Day 21. *P<0.05, **P<0.001, One way ANOVA followed by Dunnett’s test compared to Control
Discussion
We have synthesized and identified a series of hydroxamic acid derivatives designed to inhibit HDAC resulting in anti-cancer activity. In vitro mechanism of action studies demonstrate that the compounds are able to inhibit HDAC with nanomolar potency and activate apoptosis pathways such as caspaze-3 enzyme activation and cause cell death in a wide range of cancer cell lines. Four compounds PAT-1101, PAT-1103, PAT-1118 and PAT-1125 that were identified from the primary screening were tested against a panel of cancer cell lines. All the compounds exhibited anti-proliferative activity against the cancer cell types, with greater or similar potency than that of leading HDAC inhibitors in development. The compounds being novel HDAC inhibitors with good cytotoxic activity against a variety of Human tumor cell lines. Studies to evaluate the drug-likeness of the compounds such as pharmacokinetic profiling and in vivo safety studies would help in developing them as anti-cancer drugs. Improved bioavailability and safety profile of these compounds would help in determining their potential to be more effective in clinical trials than other HDAC inhibitors with poor pharmacokinetic properties and dose limiting side effects.
Acknowledgements
The Authors sincerely thank the management of Anthem Biosciences for their constant support and encouragement in carrying out this study.
Conflict of interest: None
References
- Hildmann C, Riester D, Schwienhorst A (2007) Histone deacetylases–an important class of cellular regulators with a variety of functions. Appl Microbiol Biotechnol 75: 487-497. [crossref]
- Witt O, Deubzer HE, Milde T, Oehme I (2009) HDAC family: What are the cancer relevant targets? Cancer Lett 277: 8-21. [crossref]
- Haberland M, Johnson A, Mokalled MH, Montgomery RL, Olson EN (2009) Genetic dissection of histone deacetylase requirement in tumor cells. Proc Natl Acad Sci U S A 106: 7751-7755. [crossref]
- Weichert W (2009) HDAC expression and clinical prognosis in human malignancies. Cancer Lett 280: 168-176. [crossref]
- Marks PA (2010) The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin Investig Drugs 19: 1049–1066.
- Mercurio C, Minucci S, and Pelicci PG (2010) Histone deacetylases and epigenetic therapies of hematological malignancies. Pharmacol Res 62:18-34.
- Stimson L, Wood V, Khan O, Fotheringham S, La Thangue NB (2009) HDAC inhibitor-based therapies and haematological malignancy. Ann Oncol 20: 1293-1302. [crossref]
- Drummond DC, Noble CO, Kirpotin DB, Guo Z, Scott GK (2005) Clinical development of histone deacetylase inhibitors as anticancer agents. Annual Review of Pharmacology and Toxicology 495–528
- Marks PA, Richon VM, Rifkind RA (2000) Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. Journal of the National Cancer Institute 15: 1210–1216.
- Prince HM, Bishton MJ, Harrison SJ (2009) Clinical studies of histone deacetylase inhibitors. Clinical Cancer Research 12: 3958–3969.
- Marks PA, Breslow R (2007) Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nature Biotechnology 25: 84–90.
- Piekarz RL, Frye R, Turner M, et al. (2009) Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. Journal of Clinical Oncology 32: 5410–5417.
- Elaut G, Rogiers V, Vanhaecke T (2007)The pharmaceutical potential of histone deacetylase inhibitors. Curr Pharm Des 13: 2584-2620. [crossref]
- Das, Jagattaran, Natesan, Selvakumar; Trehan, et al. (2003) Preparation of triazolyl-phenyl substituted morpholine/thiomorpholine derivatives as antibacterial agents. PCT Int Appl 2003059894.
- Dennis Wegener, Christian Hildmann, Daniel Riester, Andreas Schober, Franz-Josef Meyer-Almes, et al. (2008) Identification of novel small-molecule histone deacetylase inhibitors by medium-throughput screening using a fluorigenic assay. Biochemical Journal J 413:143-150;