Abstract
Introduction: This phase I multiple ascending dose study investigated the safety, pharmacokinetic, and pharmacodynamic properties of zalfermin in healthy participants.
Methods: Male participants aged 22–55 years and female participants aged 22–45 years with a body mass index between 27.0–39.9 kg/m2 were randomized 3:1 to receive either once-weekly subcutaneous zalfermin in multiple ascending doses of 3 mg, 9 mg, 27 mg, 60 mg, and 120 mg, or placebo for 12 weeks. Blood samples were obtained for endpoint assessments. The primary endpoint was the total number of treatment-emergent adverse events (TEAEs) from treatment administration to end of follow-up.
Results: Overall, 57 participants were enrolled. A total of 237 TEAEs were reported in 87.7% of participants; these were all mild to moderate in severity. TEAEs were mainly gastrointestinal-related and most prevalent in the 120 mg group; thus, treatment at this dose level was terminated prematurely. Dose proportionality was established for the maximum concentration of zalfermin in serum at steady state, and the geometric mean for the time to maximum concentration of zalfermin in serum at steady state ranged from 29–38 hours. The plasma half-life of zalfermin was approximately 120 hours and significant improvements in plasma lipid profiles were observed. The maximum tolerated (multiple ascending) dose of zalfermin was 60 mg and was compatible with once-weekly dosing.
Conclusions: The pharmacodynamic profile of zalfermin, particularly the observed improvement in lipids, is promising for the treatment of a range of cardiometabolic diseases including metabolic dysfunction-associated steatohepatitis. Further clinical development and investigations into zalfermin are warranted.
Keywords
Pharmacokinetics, Pharmacodynamics, Phase I, Safety, Tolerance
Introduction
Metabolic dysfunction-associated steatohepatitis (MASH) is a progressive form of metabolic dysfunction-associated steatotic liver disease (MASLD) [1], characterized by abnormal accumulation of fat in the liver (steatosis), inflammation, hepatocellular ballooning, and fibrosis [2,3]. The prevalence of both MASLD and MASH is increasing worldwide [4-6]. Approved pharmacotherapies that effectively treat MASLD and MASH alongside other cardiometabolic-related comorbidities are limited [7]; thus, there is a high unmet need for new therapies.
Fibroblast growth factor 21 (FGF21) was discovered as a metabolic regulator in 2005 [8]. Based on preclinical trials investigating obesity- related metabolic conditions, activation of the FGF21 receptor complex has been associated with several beneficial effects. These include sustained weight loss, reduced blood glucose and triglyceride (TG) levels, improvements in insulin sensitivity and hepatic steatosis [9], reduced low-density lipoprotein cholesterol (LDL-C) levels, and increased high-density lipoprotein cholesterol (HDL-C) levels [10-12]. This has led to the pursuit of FGF21 as a potential pharmacotherapy for the treatment of MASH and other metabolic-related conditions [13,14]. Zalfermin is a long-acting, proteolytically stabilized FGF21 analog, which has been preclinically shown to induce pharmacologically mediated weight loss accompanied by an improved serum lipid profile [15].
In a first-in-human, single ascending dose (SAD) study investigating the safety, pharmacokinetics (PK), and pharmacodynamics (PD) of zalfermin in healthy males, zalfermin, administered in the dose range of 2 to 180 mg, was found to have an acceptable safety profile regardless of participants’ race and ethnicity, a plasma half-life of approximately 120 hours, and demonstrated improvements in plasma lipid profiles [16]. However, to date, zalfermin has not been tested in a human female population nor via a repeated dosing regimen.
Given the long plasma half-life of zalfermin and improvement in the plasma lipid profile observed in the SAD study [16], this phase I trial investigated the safety, tolerability, PK, and PD of zalfermin when administered multiple times weekly in a healthy male and female population.
Furthermore, literature has shown that increases in FGF21 can cause a decrease in the luteinizing hormone surge and female fertility in mice [17-19]; therefore, zalfermin may mediate a reversible pause in the menstrual cycle, potentially contributing to hypothalamic amenorrhea. Thus, we collected reproductive hormone and menstrual cycle data from female participants to evaluate this.
Materials and Methods
Study Design and Participants
This was a phase I, randomized, double-blind, placebo-controlled trial (NCT03479892) conducted at a single clinical research site that enrolled female participants (aged 22–45 years) and male participants (aged 22–55 years) with overweight or obesity (body mass index 27.0–39.9 kg/m2) who were otherwise healthy. All participants were required to be generally healthy as judged by the investigator based on medical history; physical examination; and the results of vital signs, electrocardiogram, and clinical laboratory tests performed during the screening visit.
Based on nonclinical findings and previous literature [15,17-20], the menstrual cycle and reproductive hormones were monitored, and a pelvic ultrasound scan was conducted for female participants. Female participants were required to have regular menstrual cycles (defined as 24–35 days between the first day of menses and the end of the cycle, for the two most recent menstrual periods; this was self- reported), and have bilateral tubal ligation or bilateral salpingectomy or use a nonhormonal intrauterine device.
Key exclusion criteria for all participants included any clinically significant disease history and use of any prescription or non- prescription medication. The full list of exclusion criteria is in the supplementary appendix.
Participants were randomized in a 3:1 ratio to receive either once-weekly subcutaneous zalfermin in multiple ascending doses (MADs) of 3, 9, 27, 60, and 120 mg or placebo for 12 weeks. Sequential initiation was included to allow for safety clearance evaluation within each cohort before proceeding to the next cohort. The trial design can be found in Figure S1.
Safety Outcomes
The primary endpoint was the total number of treatment-emergent adverse events from the time of first zalfermin administration at baseline (day 1) to the end of the follow-up period (day 112). A treatment-emergent adverse event was defined as any event that either had onset after administration of the trial product but no later than the follow-up visit or was present before the trial product was administered and increased in severity during the treatment period but no later than the follow-up visit. Stopping criteria are listed in the supplementary appendix. Hereafter, treatment-emergent adverse events will be referred to as adverse events (AEs).
Secondary safety endpoints were changes from baseline to follow- up in vital signs, clinical laboratory safety parameters (biochemistry, hematology, and coagulation), electrocardiogram parameters, number of injection site reactions, and the presence of anti-zalfermin antibodies. Additional exploratory safety endpoints are listed in the supplementary appendix.
Supportive Secondary PK Endpoints
Blood samples for the PK analysis of zalfermin were collected from each participant on days 1 and 2 at baseline and at the start of the treatment period. Further blood samples were taken on days 8, 15, 22, 36, 50, 64, 78, 79, and 80 during the treatment phase; on day 85 (end of treatment [EOT]); days 92, 99, and 106 post-treatment; and on day 112 (follow-up). Parameters assessed were the terminal serum half-life of zalfermin at steady state (t½,SS), the maximum zalfermin serum concentration at steady state (Cmax, SS), the time to maximum zalfermin serum concentration at steady state (tmax, SS), and the apparent total serum clearance of zalfermin at steady state (CL/FSS). Serum zalfermin concentrations were measured from last dose (day 78, pre-dose) until follow-up (day 112). Exploratory PK parameters are listed in the supplementary appendix.
PK Sampling
The bioanalysis of zalfermin was performed by the Department of Development Bioanalysis, Novo Nordisk A/S. Zalfermin was assessed in serum by a validated enzyme-linked immunosorbent assay according to departmental procedures, the US Food and Drug Administration 2001 guidance on validation of bioanalytical methods [21], and the European Medicines Agency guideline on bioanalytical methods validation and current practice [22]. The lower limit of quantification for the assay was defined as 1.00 nmol/L. However, to avoid positive pre-dose samples, the lower limit of quantification was raised from 1.00 nmol/L (version 2.0) to 2.00 nmol/L.
Study samples were analyzed using an analyte-specific capture antibody and a second (detection) analyte-specific biotin- labeled antibody. The antibody–antigen complex was visualized by 3,3’,5,5’-Tetramethylbenzidine substrate. Quantification was performed using the optical density values at 450 nm (reference wavelength at 620 nm was subtracted). All calibration standards and unknown samples were analyzed in duplicate determinations (2 wells), and quality control samples were analyzed in two duplicates (2 × 2 wells). Sunrise (Tecan), controlled by Magellan version 7.1, was the instrument software used and Thermo Scientific Corporation Watson™ Bioanalytical LIMS version 7.4.2 (also called DReS), was the computer application software used for the testing systems.
Exploratory PD Endpoints and Sampling
Body weight, waist circumference, whole body fat mass, and whole body lean mass were assessed from baseline (day 1) to EOT (day 85). Blood samples were obtained for the analysis of TG, total cholesterol (TC), HDL-C, LDL-C, very low-density lipoprotein cholesterol (VLDL-C), and beta-hydroxybutyrate levels from baseline to EOT. Estimated treatment ratios (ETRs) for zalfermin treatment groups versus placebo were calculated for all lipids. Blood samples were also taken for the analyses of leptin and soluble leptin receptor, fasting FGF21, and glucose metabolism parameters (fasting serum glucose [FSG], fasting serum insulin [FSI], fasting plasma glucagon [FPG], and glycated hemoglobin [HbA1c]) from baseline to EOT. ETRs for zalfermin treatment groups versus placebo were calculated for hormones. Changes in insulin and glucose area under the curves (AUCs) related to the oral glucose tolerance test (OGTT) from baseline to EOT were also assessed as exploratory endpoints, and ETRs for zalfermin treatment groups versus placebo were also calculated for the OGTT parameters.
Statistical Analyses
Safety endpoints were analyzed using the safety analysis set (SAS; all participants who had been exposed to ≥1 dose of the trial product). Participants in the SAS contributed to the evaluation ‘as treated’. AEs were summarized using descriptive statistics. All AEs were coded using Medical Dictionary for Regulatory Activities (MedDRA) version 20.1. The statistical analyses for the secondary and exploratory safety endpoints are listed in the supplementary appendix.
PK and PD endpoints were analyzed using the full analysis set (including all randomized participants who received ≥1 dose of the trial product). Participants in the full analysis set contributed to the evaluation ‘as treated’. Further details on the statistical analyses for the supportive secondary PK endpoints are described in the supplementary appendix. All PD endpoints were summarized by zalfermin dose and placebo using descriptive statistics. Further details on the statistical analyses for the PD endpoints are described in the supplementary appendix.
Results
Baseline Characteristics
Overall, 57 participants were enrolled with a mean age (standard deviation [SD]) of 37.9 (6.6) years. The majority were male (54.4%) and White (80.7%). Mean (SD) body weight and body mass index were 93.8 (14.1) kg and 32.5 (3.2) kg/m2, respectively (Table 1). Mean (SD) fasting FGF21 in plasma was 240.2 (248.9) pg/mL. The full list of baseline characteristics is presented in Table 1.
Table 1: Baseline characteristics
|
Characteristic |
Zalfermin | ||||||
| 3 mg(n = 10) | 9 mg(n = 9) | 27 mg(n = 9) | 60 mg(n = 9) | 120 mg(n = 6) | Placebo(n = 14) |
Total (N = 57) |
|
| Age, years |
37.2 (6.1) |
36.2 (4.4) | 39.2 (5.4) | 34.1 (8.6) | 41.5 (5.3) | 39.4 (7.4) |
37.9 (6.6) |
| Sex, n (%) | |||||||
|
Male |
3 (30.0) |
3 (33.3) | 5 (55.6) | 6 (66.7) | 6 (100.0) | 8 (57.1) | 31 (54.4) |
|
Female |
7 (70.0) | 6 (66.7) | 4 (44.4) | 3 (33.3) | 0 (0.0) | 6 (42.9) |
26 (45.6) |
| Female of childbearing potential | |||||||
|
Yes |
4 (57.1) |
4 (66.7) | 3 (75.0) | 2 (66.7) | 0 (0.0) | 2 (33.3) | 15 (57.7) |
|
No |
3 (42.9) | 2 (33.3) | 1 (25.0) | 1 (33.3) | 0 (0.0) | 4 (66.7) |
11 (42.3) |
| Race, n (%) | |||||||
|
White |
8 (80.0) |
7 (77.8) | 9 (100.0) | 7 (77.8) | 4 (66.7) | 11 (78.6) | 46 (80.7) |
|
Asian |
0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
0 (0.0) |
|
Black or African American |
2 (20.0) |
2 (22.2) | 0 (0.0) | 2 (22.2) | 1 (16.7) | 3 (21.4) | 10 (17.5) |
|
American Indian or Alaska Native |
0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (16.7) | 0 (0.0) |
1 (1.8) |
| Body weight, kg |
89.1 (10.8) |
89.4 (17.8) | 98.8 (13.6) | 97.2 (19.9) | 99.1 (9.5) | 92.1 (10.9) | 93.8 (14.1) |
|
BMI, kg/m2 |
32.5 (2.5) | 32.1 (4.0) | 34.1 (2.7) | 33.7 (4.1) | 32.1 (3.7) | 31.2 (2.5) |
32.5 (3.2) |
| Waist circumference, cm |
103.2 (6.2) |
103.9 (13.1) | 108.1 (8.9) | 106.9 (12.8) | 108.4 (11.4) | 101.3 (11.0) | 104.7 (10.6) |
|
Triglycerides, mg/dL |
120 (41) | 224 (110) | 162 (56) | 180 (147) | 199 (39) | 124 (59) |
162 (89) |
| LDL cholesterol, mg/dL |
116 (34) |
126 (23) | 123 (23) | 153 (39) | 162 (31) | 129 (37) | 133 (34) |
|
HDL cholesterol, mg/dL |
57 (17) | 45 (9) | 51 (12) | 45 (7) | 40 (6) | 50 (12) |
49 (12) |
| FGF21 in plasma, pg/mL |
310.1 (285.5) |
306.6(277.2) | 310.4(329.3) | 257.7(310.3) | 136.8(47.0) | 135.6(95.6) |
240.2 (248.9) |
| HbA1c, % |
5.2 (0.3) |
5.3 (0.2) | 5.4 (0.3) | 5.3 (0.3) | 5.6 (0.3) | 5.4 (0.4) |
5.3 (0.3) |
Data are mean (standard deviation) unless otherwise stated.
BMI, body mass index; FGF21, fibroblast growth factor 21; HbA1c, glycated hemoglobin; HDL, high-density lipoprotein; LDL, low-density lipoprotein.
Safety and Tolerability
Primary Endpoint AEs
In total, 237 AEs were reported for 50 participants (87.7%), and were mild to moderate in severity and transient (Table 2). Of the 237 events, 204 AEs in 40 participants were reported across the five zalfermin treatment groups, and 33 events in 10 participants were reported in the placebo group. The highest number of AEs was reported in the 9 mg treatment group, with 61 events in nine participants; no clear dose dependency in the number of AEs was observed (Table 2). AEs were mainly gastrointestinal (GI)-related, with 49 events recorded in 27 participants across all treatment groups including placebo. GI- related AEs were most prevalent in the zalfermin 120 mg treatment group, with four events of vomiting recorded in four participants and six events of nausea recorded in five participants (Table 2). As a result of this, treatment with the 120 mg dose was terminated prematurely. Overall, the most frequently reported AEs by preferred term were increased appetite (Table 2) and nausea, and injection site reactions.
Five participants withdrew from the trial and five participants discontinued treatment due to AEs (Table 2). A total of 134 AEs in 42 participants were assessed to have probable or possible relation to the trial product (Table 2). No deaths, serious AEs, or AEs related to technical complaints were reported.
Table 2: AEs from time of first zalfermin administration (day 1) to the end of the follow-up period (day 112).
| Zalfermin | |||||||
| 3 mg(n = 10) | 9 mg(n = 9) | 27 mg(n = 9) | 60 mg(n = 9) | 120 mg(n = 6) | Placebo (n = 14) |
Total (N = 57) |
|
|
n (%) | E |
n (%) | E | n (%) | E | n (%) | E | n (%) | E | n (%) | E |
n (%) | E |
|
| AEs |
8 (80.0) | 23 |
9 (100.0) | 61 | 8 (88.9) | 42 | 9 (100.0) | 50 | 6 (100.0) | 28 | 10 (71.4) | 33 | 50 (87.7) | 237 |
|
Serious AEs |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 |
0 (0.0) | 0 |
| AEs leading to withdrawal |
0 (0.0) | 0 |
2 (22.2) | 2 | 0 (0.0) | 0 | 1 (11.1) | 1 | 2 (33.3) | 2 | 0 (0.0) | 0 | 5 (8.8) | 5 |
|
AEs leading to treatment discontinuation |
0 (0.0) | 0 | 3 (33.3) | 3 | 0 (0.0) | 0 | 2 (22.2) | 2 | 0 (0.0) | 0 | 0 (0.0) | 0 |
5 (8.8) | 5 |
| Related to trial product | |||||||
|
Probable |
5 (50.0) | 9 |
7 (77.8) | 21 | 7 (77.8) | 22 | 8 (88.9) | 18 | 5 (83.3) | 12 | 4 (28.6) | 5 | 36 (63.2) | 87 |
|
Possible |
4 (40.0) | 7 | 5 (55.6) | 10 | 3 (33.3) | 3 | 4 (44.4) | 10 | 4 (66.7) | 7 | 6 (42.9) | 10 |
26 (45.6) | 47 |
|
Unlikely |
7 (70.0) | 7 |
9 (100.0) | 30 | 6 (66.7) | 17 | 8 (88.9) | 22 | 4 (66.7) | 9 | 9 (64.3) | 18 |
43 (75.4) | 103 |
| Severity | |||||||
|
Severe |
0 (0.0) | 0 |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 |
|
Moderate |
5 (50.0) | 5 | 7 (77.8) | 20 | 5 (55.6) | 8 | 7 (77.8) | 17 | 5 (83.3) | 15 | 8 (57.1) | 9 |
37 (64.9) | 74 |
|
Mild |
8 (80.0) | 18 |
9 (100) | 41 | 8 (88.9) | 34 | 9 (100) | 33 | 6 (100) | 13 | 10 (71.4) | 24 | 50 (87.7) | 163 |
|
Injection site reactions |
1 (10.0) | 2 | 4 (44.4) | 10 | 6 (66.7) | 18 | 5 (55.6) | 11 | 0 (0.0) | 0 | 2 (14.3) | 3 |
18 (31.6) | 44 |
| Symptoms | |||||||
|
Itching |
1 (100.0) | 1 |
2 (50.0) | 4 | 2 (33.3) | 5 | 2 (40.0) | 2 | 0 (0.0) | 0 | 0 (0.0) | 0 | 7 (38.9) | 12 |
|
Redness |
1 (100.0) | 1 | 4 (100.0) | 4 | 5 (83.3) | 11 | 3 (60.0) | 3 | 0 (0.0) | 0 | 0 (0.0) | 0 |
13 (72.2) | 19 |
|
Ecchymosis |
0 (0.0) | 0 |
1 (25.0) | 1 | 1 (16.7) | 1 | 2 (40.0) | 3 | 0 (0.0) | 0 | 2 (100.0) | 3 | 6 (33.3) | 8 |
|
Related to technical complaint |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 |
0 (0.0) | 0 |
| AEs by system organ class and MedDRA Preferred Term | |||||||
| Metabolism and nutrition disorders |
5 (50.0) | 5 |
6 (66.7) | 8 | 5 (55.6) | 6 | 9 (100.0) | 10 | 1 (16.7) | 1 | 4 (28.6) | 4 | 30 (52.6) | 34 |
|
Increased appetite |
5 (50.0) | 5 | 5 (55.6) | 6 | 5 (55.6) | 5 | 8 (88.9) | 8 | 1 (16.7) | 1 | 4 (28.6) | 4 |
28 (49.1) | 29 |
|
Glucose tolerance impaired |
0 (0.00) | 0 |
1 (11.1) | 1 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 2 (3.5) | 2 |
|
Hyperphagia |
0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 |
1 (1.8) | 1 |
|
Lack of satiety |
0 (0.0) | 0 |
0 (0.0) | 0 | 0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
Type 2 diabetes mellitus |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 |
1 (1.8) | 1 |
| Gastrointestinal disorders |
3 (30.0) | 3 |
6 (66.7) | 9 | 3 (33.3) | 6 | 3 (33.3) | 7 | 6 (100.0) | 15 | 6 (42.9) | 9 | 27 (47.4) | 49 |
|
Vomiting |
0 (0.0) | 0 | 2 (22.2) | 2 | 2 (22.2) | 2 | 3 (33.3) | 3 | 4 (66.7) | 4 | 0 (0.0) | 0 |
11 (19.3) | 11 |
|
Nausea |
0 (0.0) | 0 |
3 (33.3) | 3 | 1 (11.1) | 1 | 1 (11.1) | 2 | 5 (83.3) | 6 | 0 (0.0) | 0 | 10 (17.5) | 12 |
|
Diarrhea |
1 (10.0) | 1 | 1 (11.1) | 1 | 0 (0.0) | 0 | 2 (22.2) | 2 | 2 (33.3) | 2 | 1 (7.1) | 1 |
7 (12.3) | 7 |
|
Abdominal distension |
1 (10.0) | 1 |
0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 2 (14.3) | 2 | 4 (7.0) | 4 |
|
Lower abdominal pain |
1 (10.0) | 1 | 2 (22.2) | 2 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 |
3 (5.3) | 3 |
|
Gastro-esophageal reflux disease |
0 (0.0) | 0 |
1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 2 (33.3) | 2 | 0 (0.0) | 0 | 3 (5.3) | 3 |
|
Abdominal pain |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 2 (14.3) | 2 |
2 (3.5) | 2 |
|
Flatulence |
0 (0.0) | 0 |
0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (7.1) | 1 | 2 (3.5) | 2 |
|
Upper abdominal pain |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (7.1) | 1 |
1 (1.8) | 1 |
|
Constipation |
0 (0.0) | 0 |
0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
Dry mouth |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (7.1) | 1 |
1 (1.8) | 1 |
|
Food poisoning |
0 (0.0) | 0 |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (7.1) | 1 | 1 (1.8) | 1 |
|
Retching |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (16.7) | 1 | 0 (0.0) | 0 |
1 (1.8) | 1 |
| Investigations |
4 (40.0) | 4 |
6 (66.7) | 6 | 3 (33.3) | 4 | 3 (33.3) | 7 | 2 (33.3) | 2 | 4 (28.6) | 7 | 22 (38.6) | 30 |
|
Weight increased |
3 (30.0) | 3 | 4 (44.4) | 4 | 2 (22.2) | 2 | 2 (22.2) | 2 | 0 (0.0) | 0 | 1 (7.1) | 1 |
12 (21.1) | 12 |
|
DBP increased |
0 (0.0) | 0 |
0 (0.0) | 0 | 0 (0.0) | 0 | 2 (22.2) | 3 | 1 (16.7) | 1 | 0 (0.0) | 0 | 3 (5.3) | 4 |
|
ALT increased |
0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (7.1) | 1 |
2 (3.5) | 2 |
|
Blood creatine phosphokinase increased |
0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (7.1) | 1 |
2 (3.5) | 2 |
|
CRP increased |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (11.1) | 1 | 1 (16.7) | 1 | 0 (0.0) | 0 |
2 (3.5) | 2 |
|
AST increased |
0 (0.0) | 0 |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (7.1) | 1 | 1 (1.8) | 1 |
|
Blood glucose decreased |
0 (0.0) | 0 |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (7.1) | 1 | 1 (1.8) | 1 |
|
Blood pressure increased |
0 (0.0) | 0 | 0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 |
1 (1.8) | 1 |
|
SBP increased |
0 (0.0) | 0 |
0 (0.0) | 0 | 0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
Blood triglycerides increased |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (7.1) | 1 |
1 (1.8) | 1 |
|
GFR decreased |
1 (10.0) | 1 |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
LDL cholesterol increased |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (7.1) | 1 |
1 (1.8) | 1 |
| General disorders and administration site conditions |
2 (20.0) | 3 |
5 (55.6) | 12 | 6 (66.7) | 19 | 5 (55.6) | 13 | 0 (0.0) | 0 | 3 (21.4) | 4 | 21 (36.8) | 51 |
|
Injection site erythema |
1 (10.0) | 1 | 4 (44.4) | 5 | 5 (55.6) | 11 | 3 (33.3) | 3 | 0 (0.0) | 0 | 0 (0.0) | 0 |
13 (22.8) | 20 |
|
Injection site hemorrhage |
0 (0.0) | 0 |
1 (11.1) | 1 | 1 (11.1) | 1 | 2 (22.2) | 3 | 0 (0.0) | 0 | 2 (14.3) | 3 | 6 (10.5) | 8 |
|
Injection site pruritus |
1 (10.0) | 1 | 2 (22.2) | 4 | 2 (22.2) | 5 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 |
6 (10.5) 11 |
|
Early satiety |
0 (0.0) | 0 |
0 (0.0) | 0 | 1 (11.1) | 1 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 2 (3.5) | 2 |
|
Fatigue |
1 (10.0) | 1 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 |
2 (3.5) | 2 |
|
Asthenia |
0 (0.0) | 0 |
1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
Injection site bruising |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 |
1 (1.8) | 1 |
|
Injection site hematoma |
0 (0.0) | 0 |
0 (0.0) | 0 | 0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
Injection site induration |
0 (0.0) | 0 | 0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 |
1 (1.8) | 1 |
|
Injection site edema |
0 (0.0) | 0 |
0 (0.0) | 0 | 0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
Injection site reaction |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 |
1 (1.8) | 1 |
|
Non-cardiac chest pain |
0 (0.0) | 0 |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (7.1) | 1 | 1 (1.8) | 1 |
|
Suprapubic pain |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 |
1 (1.8) | 1 |
| Infections and infestations |
1 (10.0) | 1 |
2 (22.2) | 5 | 2 (22.2) | 2 | 5 (55.6) | 7 | 3 (50.0) | 4 | 2 (14.3) | 2 | 15 (26.3) | 21 |
|
Upper respiratory tract infection |
0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 3 (33.3) | 3 | 0 (0.0) | 0 | 1 (7.1) | 1 |
5 (8.8) | 5 |
|
Gastroenteritis |
0 (0.0) | 0 |
0 (0.0) | 0 | 0 (0.0) | 0 | 2 (22.2) | 2 | 1 (16.7) | 1 | 1 (7.1) | 1 | 4 (7.0) | 4 |
|
Viral upper respiratory tract infection |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 3 (50.0) | 3 | 0 (0.0) | 0 |
3 (5.3) | 3 |
|
Urinary tract infection |
0 (0.0) | 0 |
1 (11.1) | 1 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 2 (3.5) | 2 |
|
Anal abscess |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 |
1 (1.8) | 1 |
|
Chlamydial infection |
1 (10.0) | 1 |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
Cystitis |
0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 |
1 (1.8) | 1 |
|
Ear infection |
0 (0.0) | 0 |
1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
Lower respiratory tract infection |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 |
1 (1.8) | 1 |
|
Rhinitis |
0 (0.0) | 0 |
0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
Vulvovaginal mycotic infection |
0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 |
1 (1.8) | 1 |
| Nervous system disorders |
1 (10.0) | 1 |
2 (22.2) | 2 | 2 (22.2) | 2 | 1 (11.1) | 1 | 2 (33.3) | 2 | 2 (14.3) | 2 | 10 (17.5) | 10 |
|
Headache |
0 (0.0) | 0 | 2 (22.2) | 2 | 2 (22.2) | 2 | 1 (11.1) | 1 | 1 (16.7) | 1 | 2 (14.3) | 2 |
8 (14.0) | 8 |
|
Dizziness |
1 (10.0) | 1 |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
Lethargy |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (16.7) | 1 | 0 (0.0) | 0 |
1 (1.8) | 1 |
| Reproductive system and breast disorders |
3 (30.0) | 4 |
2 (22.2) | 5 | 0 (0.0) | 0 | 1 (11.1) | 2 | 0 (0.0) | 0 | 1 (7.1) | 2 | 7 (12.3) | 13 |
|
Polymenorrhea |
3 (30.0) | 3 | 1 (11.1) | 1 | 0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 1 (7.1) | 1 |
6 (10.5) | 6 |
|
Hypomenorrhea |
1 (10.0) | 1 |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (7.1) | 1 | 2 (3.5) | 2 |
|
Menorrhagia |
0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 |
1 (1.8) | 1 |
|
Irregular menstruation |
0 (0.0) | 0 |
1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
Metrorrhagia |
0 (0.0) | 0 | 1 (11.1) | 2 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 |
1 (1.8) | 2 |
|
Vaginal hemorrhage |
0 (0.0) | 0 |
0 (0.0) | 0 | 0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 |
1 (1.8) | 1 |
|
Respiratory, thoracic, and mediastinal disorders |
0 (0.0) | 0 |
2 (22.2) | 2 | 2 (22.2) | 2 | 2 (22.2) | 2 | 1 (16.7) | 1 | 0 (0.0) | 0 |
7 (12.3) | 7 |
|
Cough |
0 (0.0) | 0 |
1 (11.1) | 1 | 0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 2 (3.5) | 2 |
|
Allergic rhinitis |
0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 |
2 (3.5) | 2 |
|
Dyspnea |
0 (0.0) | 0 |
0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
Rhinorrhea |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (16.7) | 1 | 0 (0.0) | 0 |
1 (1.8) | 1 |
|
Upper respiratory tract congestion |
0 (0.0) | 0 |
0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
Skin and subcutaneous tissue disorders |
0 (0.0) | 0 | 4 (44.4) | 4 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (7.1) | 1 |
5 (8.8) | 5 |
|
Hyperhidrosis |
0 (0.0) | 0 |
3 (33.3) | 3 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 3 (5.3) | 3 |
|
Pruritus |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (7.1) | 1 |
1 (1.8) | 1 |
|
Urticaria |
0 (0.0) | 0 |
1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
Injury, poisoning, and procedural complications |
0 (0.0) | 0 | 3 (33.3) | 3 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (16.7) | 1 | 0 (0.0) | 0 |
4 (7.0) | 4 |
|
Contusion |
0 (0.0) | 0 |
2 (22.2) | 2 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 2 (3.5) | 2 |
|
Injection-related reaction |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (16.7) | 1 | 0 (0.0) | 0 |
1 (1.8) | 1 |
|
Injury |
0 (0.0) | 0 |
1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
Musculoskeletal and connective tissue disorders |
0 (0.0) | 0 | 2 (22.2) | 2 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 |
3 (5.3) | 3 |
|
Back pain |
0 (0.0) | 0 |
1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
Musculoskeletal discomfort |
0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 |
1 (1.8) | 1 |
|
Tendonitis |
0 (0.0) | 0 |
0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
Blood and lymphatic system disorders |
0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 |
2 (3.5) | 2 |
|
Anemia |
0 (0.0) | 0 |
1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
Lymphadenopathy |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 |
1 (1.8) | 1 |
| Psychiatric disorders |
0 (0.0) | 0 |
1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (7.1) | 1 | 2 (3.5) | 2 |
|
Insomnia |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (7.1) | 1 |
1 (1.8) | 1 |
|
Decreased libido |
0 (0.0) | 0 |
1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
Cardiac disorders |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (16.7) | 1 | 0 (0.0) | 0 |
1 (1.8) | 1 |
|
Postural orthostatic tachycardia syndrome |
0 (0.0) | 0 |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (16.7) | 1 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
Ear and labyrinth disorders |
0 (0.0) | 0 | 1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 |
1 (1.8) | 1 |
|
Ear discomfort |
0 (0.0) | 0 |
1 (11.1) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
Eye disorders |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (16.7) | 1 | 0 (0.0) | 0 |
1 (1.8) | 1 |
|
Cataract |
0 (0.0) | 0 |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (16.7) | 1 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
Immune system disorders |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (7.1) | 1 |
1 (1.8) | 1 |
|
Allergy to chemicals |
0 (0.0) | 0 |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (7.1) | 1 | 1 (1.8) | 1 |
|
Neoplasms benign, malignant, and unspecified (including cysts and polyps) |
1 (10.0) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 |
1 (1.8) | 1 |
|
Uterine leiomyoma |
1 (10.0) | 1 |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 1 (1.8) | 1 |
|
Renal and urinary disorders |
1 (10.0) | 1 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 |
1 (1.8) | 1 |
|
Hematuria |
1 (10.0) | 1 |
0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 | 0 (0.0) | 0 |
1 (1.8) | 1 |
Treatment with the 120 mg dose was discontinued early due to AEs.
AE, adverse event; ALT, alanine aminotransferase; AST, aspartate aminotransferase; CRP, C-reactive protein; DBP, diastolic blood pressure; E, number of events; GFR, glomerular filtration rate; LDL, low-density lipoprotein; MedDRA, Medical Dictionary for Regulatory Activities; SBP, systolic blood pressure.
Supportive Secondary Safety Endpoints
Vital Signs and ECG
Administration of zalfermin was not associated with any clinically relevant changes in vital signs such as body temperature, pulse, or respiratory rate (Table S1). Slight increases in systolic blood pressure (SBP) and diastolic blood pressure (DBP) were observed in participants who received the two highest doses of zalfermin; one AE of increased SBP in the zalfermin 60 mg treatment group and four AEs of increased DBP (three in the 60 mg treatment group, one in the 120 mg treatment group; Table 2). No AEs of increased heart rate were reported at follow-up (Table S1).
Clinical Laboratory Parameters
In total, six AEs related to clinical laboratory parameters across the zalfermin treatment groups were reported (ie, glomerular filtration rate decrease [n = 1], blood creatine phosphokinase increase [n = 1], alanine aminotransferase increase [n = 1], blood cholesterol increase [n = 1], and C-reactive protein increase [n = 2]); these events were deemed to be unlikely related to the trial product, and all were mild to moderate in severity.
Injection Site Reactions
In total, 44 injection site reactions were reported in 18 participants (31.6%), but did not appear to be dose dependent, with the highest number of events (6) reported in the zalfermin 27 mg dose (Table 2). All injection site reactions were mild; included mainly redness, ecchymosis, and itching; and duration ranged between 16 minutes and 13 days. The dose volume injected per dose is presented in Table S2.
Anti-zalfermin Antibodies
Across all zalfermin treatment groups, anti-zalfermin antibodies were detected in 14 participants (24.6%) post-baseline, all of whom had cross reactivity to FGF21 (Table S3). Of these 14 participants, 10 were positive for neutralizing antibodies toward FGF21 (Table S3). Presence of anti-zalfermin antibodies was not related to any AEs and they were not associated with any effects on PK.
Exploratory Safety Endpoints
Results for exploratory safety outcomes including menses assessments, pelvic ultrasound, and reproductive hormone assessments in females; bone mineral density; and patient-reported outcomes are presented in the supplementary appendix (Tables S4– S7).
Pharmacokinetics
The concentration–time profiles across zalfermin doses are presented in Figure 1. Across the zalfermin 3 mg to 60 mg treatment groups, geometric mean Cmax, SS ranged between 20.4 nmol/L and 399 nmol/L (Table 3) and geometric mean area under the serum concentration–time curve from time 0 to 168 hours at steady state (AUC0-168,SS) ranged between 2612 nmol*h/L and 53,369 nmol*h/L (Table S8). Across all zalfermin treatment groups, dose proportionality was established for Cmax,SS (2β estimate: 1.94; 95% confidence interval: 1.80–2.10; P = 0.4439) and AUC0-168,SS (estimate 1.97; 95% confidence interval: 1.83–2.12; P = 0.6712). The geometric mean t1/2,SS of zalfermin ranged between 120 and 127 hours and apparent CL/FSS ranged from 0.0347 to 0.0599 L/h for the 3 mg to 60 mg doses (Table 3). Geometric mean tmax,SS ranged between 29 and 38 hours (Table 3). Further results for the exploratory PK parameters are presented in Table S8.
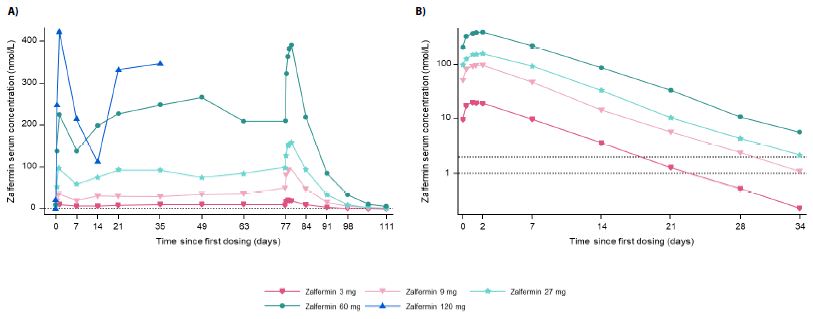
Figure 1: Pharmacokinetics of zalfermin after MADs in healthy male and female participants.
(A) shows the geometric mean for the full concentration–time profiles. (B) shows the geometric mean for concentration–time profiles at steady state and elimination on the logarithmic scale. The dotted lines in both graphs are reference lines for the LLOQ. Values below the LLOQ were imputed. LLOQ, lower limit of quantification; MAD, multiple ascending dose.
Table 3: Pharmacokinetics of zalfermin at steady state after MADs in healthy male and female participants.
| Zalfermin | ||||
| 3 mg(n = 10) | 9 mg(n = 9) | 27 mg(n = 9) |
60 mg (n = 9) |
|
| t1/2,SS, h |
122 (23.0) |
127 (21.3) | 120 (12.6) |
122 (5.2) |
| Cmax,SS, nmol/L |
20.4 (24.8) |
98.1 (31.4) | 162 (23.1) |
399 (24.1) |
| tmax,SS, h |
29 (71.9) |
38 (41.9) | 37 (30.2) |
37 (29.3) |
| CL/FSS, L/h |
0.0565 (22.1) |
0.0347 (31.9) | 0.0599 (25.1) |
0.0553 (24.8) |
Data are geometric mean (CV). Data are not shown for the 120 mg treatment group as treatment at this dose was discontinued early due to adverse events. CL/FSS, total apparent serum clearance at steady state; Cmax,SS, maximum plasma concentration at steady state; CV, coefficient of variation; MADs, multiple ascending doses; t1/2,SS, terminal serum half-life at steady state; tmax,SS, time to Cmax at steady state.
Pharmacodynamics
Body Weight, Waist Circumference, Whole Body Fat Mass, and Whole Body Lean Mass
Changes in body weight over time are shown in Figure 2. A statistically significant weight gain of 4.5 %-points (placebo-adjusted) was observed in the zalfermin 3 mg treatment group (P = 0.0008). For the zalfermin 60 mg treatment group, a weight loss of 1.9 %-points was observed; however, this was not statistically significant compared with placebo (Figure S2). No clinically relevant change or dose dependency was observed across any zalfermin dose for waist circumference, whole body fat mass, and whole body lean mass (Figure S2).
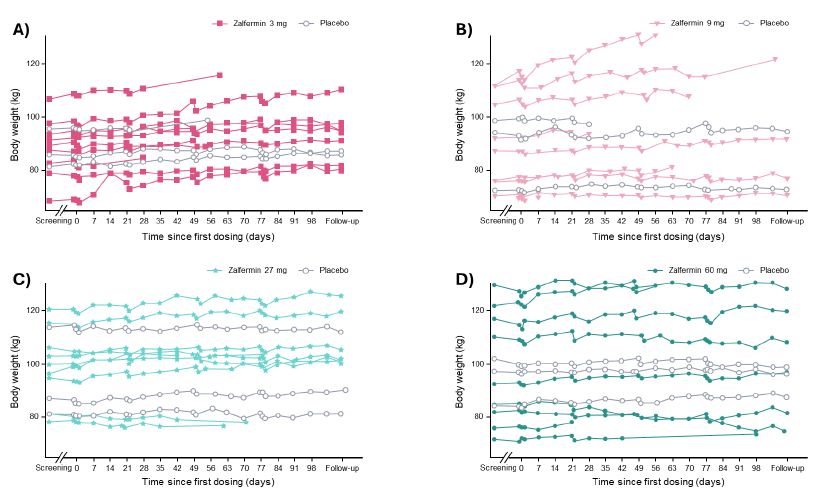
Figure 2: Change in body weight according to zalfermin dose over time: (A) 3 mg, (B) 9 mg, (C) 27 mg, and (D) 60 mg.
Data are not shown for the 120 mg treatment group as treatment at this dose was discontinued early due to adverse events.
Lipids
Sustained improvements in TG, HDL-C, and LDL-C levels were observed at EOT (Figure 3). The change from baseline to EOT in TG and VLDL-C was significant across the zalfermin 3 mg to 60 mg treatment groups versus placebo (P < 0.05) (Figure 4). Improvements in HDL-C and LDL-C were dose dependent, with the greatest improvements achieved with the zalfermin 60 mg dose versus placebo (P = 0.0113 and P = 0.0135, respectively; Figures 3 and 4). Improvements in TC were significant for the zalfermin 27 and 60 mg doses versus placebo (P = 0.0023 and P = 0.0116, respectively). For beta-hydroxybutyrate, non-significant increases favored zalfermin versus placebo with the exception of the 27 mg dose (Figure 4).
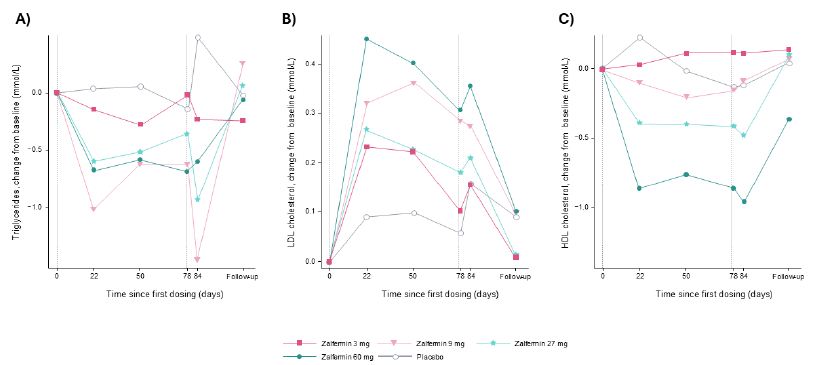
Figure 3: Change from baseline in lipids: (A) triglycerides, (B) LDL cholesterol, and (C) HDL cholesterol after MADs of zalfermin in healthy participants.
Data are not shown for the 120 mg treatment group as treatment at this dose was discontinued early due to adverse events.
Vertical dotted reference lines represent first and last doses of zalfermin.
HDL, high-density lipoprotein; LDL, low-density lipoprotein; MAD, multiple ascending dose.
Placebo-adjusted changes in the 3, 9, 27, and 60 mg treatment groups were −33%, −52%, −61%, and −48%, respectively, for TGs; −1%, −4%, −17%, and −14%, respectively, for TC; 7%, −1%, −13%, and −20%, respectively, for LDL-C; −32%, −52%, −61%, and −48%, respectively, for VLDL-C; 51%, 10%, −26%, and 12%, respectively, for beta-hydroxybutyrate; and −1%, 11%, −1%, and 20%, respectively, for HDL-C (Figure 4).
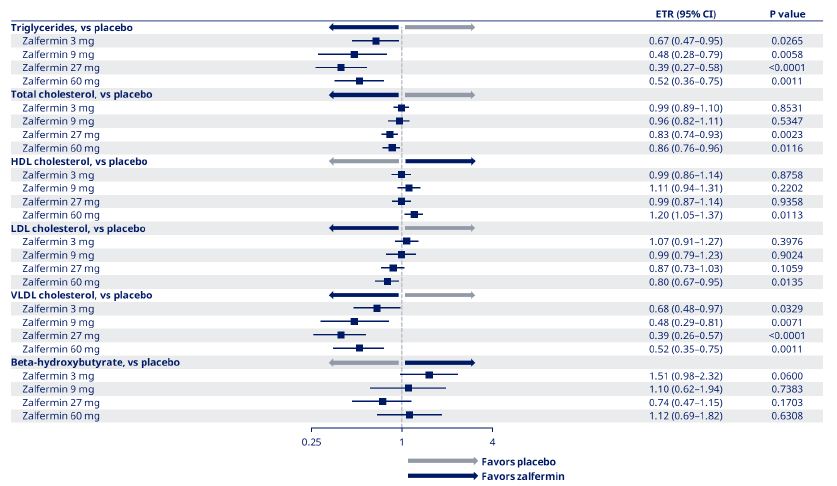
Figure 4: Estimated treatment ratios for change from baseline to end of treatment in lipids for zalfermin versus placebo.
Data are not shown for the 120 mg treatment group as treatment at this dose was discontinued early due to adverse events.
CI, confidence interval; ETR, estimated treatment ratio; HDL, high-density lipoprotein; LDL, low-density lipoprotein; VLDL, very low-density lipoprotein.
Hormones
Decreases in plasma leptin levels were observed at EOT in all zalfermin treatment groups with no clear dose dependency (Table S9). Plasma soluble leptin receptor levels increased across all the zalfermin treatment groups at EOT; changes were not dose dependent (Table S9). The ETRs for plasma leptin and plasma soluble leptin receptor levels across all zalfermin treatment groups are presented in Figure S3. Changes in plasma leptin levels favored zalfermin except for the 3 mg dose (Figure S3). Changes in plasma soluble leptin receptor levels favored zalfermin across all treatment groups (Figure S3).
Glucose Metabolism
Variations in mean glucose metabolism parameters were observed across the zalfermin treatment groups, with no clear dose dependency. No clinically meaningful changes in FSG, FSI, or FPG were observed across the zalfermin treatment groups (Table S10). A reduction in HbA1c was observed in all zalfermin treatment groups, with greatest reduction observed in the 27 and 60 mg treatment groups (Table S10).
The ETRs for the glucose AUCs were consistently but not statistically significantly slightly above 1 for the 3 mg to 60 mg treatment groups, indicating a minimally higher glucose excursion with the OGTT. This was accompanied by a slight non-significant lowering below 1 in the ETRs for insulin AUCs in the two highest treatment groups (27 and 60 mg) at EOT, compared with baseline. This tendency was not confirmed for the incremental AUCs for insulin and glucose for the highest dose of zalfermin (Table S11).
Discussion
In the current study, AEs were mainly GI related across all MADs of zalfermin and were mild to moderate in severity. Zalfermin was generally well tolerated, except for the 120 mg treatment group that was terminated early due to GI-related AEs (vomiting and nausea). No deaths, serious AEs, or AEs related to technical complaints were reported across any of the zalfermin treatment groups. Overall, zalfermin had a safety profile that was consistent with the FGF21 analog class [13,23-26]. No clinically relevant effect on the menstrual cycle, female reproductive organs, or sex hormones were observed, and no clinically relevant changes at follow-up were observed in other safety parameters. Although slight increases in SBP and DBP were observed in the higher-dose zalfermin treatment groups, these were not regarded as events directly caused by treatment but likely incidental findings. Of note, one other FGF21 analog (PF-05231023) has reported slight increases in blood pressure [27]. Low-titer anti- zalfermin antibodies were detected in 14 participants with no clinical impact. Given that the 120 mg dose in this current study was terminated prematurely, the 60 mg dose appears to be the maximum tolerated dose for weekly administration.
Dose proportionality was observed for AUC0-168,SS and Cmax,SS. The geometric mean t½,SS of zalfermin ranged from 120 to 127 hours, and tmax,SS ranged from 29 to 38 hours across the zalfermin 3 mg to 60 mg treatment groups. The approximate plasma half-life of zalfermin of 120 hours (5 days) was determined in the SAD study [16] and corroborated in this study. Therefore, zalfermin may be capable of sustained PD activity with a once-weekly dosing regimen. By comparison, other FGF21 analogs such as efruxifermin and pegozafermin have reported half-lives in humans of 3–3.5 days, which were suitable for once-weekly dosing [14,28]. Other PK parameters, for example Cmax and tmax, were also similar between this study and the SAD study [16].
Participants in the lower-dose zalfermin treatment groups experienced some weight gain, likely mediated by an increase in appetite and patient-reported changes in food preferences, which has also been described in preclinical species [29,30]. In addition, some other FGF21 analogs have shown an increase in appetite with no clinically meaningful change in body weight [13,31]. In this current study, no clinically meaningful change in body weight was observed in the 60 mg treatment group, where increase in appetite was observed, indicating a potential treatment effect on energy expenditure. By comparison, no clinically meaningful weight loss was observed in the SAD study [16]. In a previous study, efruxifermin did not demonstrate meaningful weight loss at lower doses of 7 and 21 mg compared with the higher doses of 70 and 140 mg [13]. Furthermore, in another study, efruxifermin 50 mg did show a trend toward body weight reduction [32]. Thus, it is likely that studies involving higher doses of zalfermin and of longer duration may be needed for clinically significant weight loss to be observed; however, the effect on body weight reduction would need to be carefully balanced with the observed GI-related AEs. At the time of writing, the phase II study (NCT05016882) of zalfermin 30 mg and semaglutide combination therapy is in progress to determine their effect on MASH resolution and fibrosis improvement in patients with MASH and fibrosis stages 2–4 [33]. Semaglutide has previously demonstrated glucagon-like peptide-1 receptor agonist- mediated weight reduction [34], and it is hypothesized that this would confer additional benefit alongside zalfermin treatment.
Changes in leptin favored zalfermin versus placebo across all doses (except the 3 mg dose) despite no clinically significant weight loss across zalfermin treatment groups. This may be related to the mechanism of action of zalfermin with modulation of adipokines in adipose tissue, resulting in alterations in secretion and systemic leptin sensitivity [35]. A decrease in leptin may also be related to the decrease in female fertility that was observed preclinically [15,17-20], suggesting that this may not translate to patients with overweight/ obesity.
Positive effects on the lipid profile in humans have previously been demonstrated with other FGF21 analogs [13,24-26]. The percentage changes from baseline in TG, HDL-C, LDL-C, and VLDL-C levels for the 60 mg treatment group in this analysis were –48%, 20%, –20%, and –48%, respectively. Specifically for TG and VLDL-C, significant treatment effects were observed across all zalfermin treatment groups versus placebo (3 mg to 60 mg). These results are consistent with the lipid profile improvement reported in the SAD study [16]. Although this study was conducted in healthy volunteers with overweight/ obesity, these results indicate that zalfermin may be promising not only as pharmacotherapy for the treatment of MASH but for other cardiometabolic-related disorders such as dyslipidemia and severe hypertriglyceridemia [36].
No clinically meaningful changes in FSG, FSI, and FPG were observed across the zalfermin treatment groups. This is consistent with the results from the SAD study, where no clinically relevant changes were observed in glucose metabolism parameters [16]. This could be explained by the fact that in both the current study and the SAD study, analyses were conducted in populations without diabetes, and therefore, clinically meaningful changes in glucose metabolism parameters were not expected. However, in this current analysis, reduction of HbA1c was observed and was greatest in the zalfermin 27 and 60 mg treatment groups. This is likely attributable to the MAD design of this study, with participants having a higher exposure to zalfermin compared with the SAD study [16], and where an effect on HbA1c was harder to observe with once-weekly single dosing. In addition, participants in this current study had obesity and therefore may also have been insulin resistant. Furthermore, the results of the OGTT, with a slight increase in glucose excursion rate across all doses of zalfermin and a lowering of insulin, could suggest a reduction of glucose tolerance. This finding is not supported by the HbA1c results where a reduction was observed at the higher zalfermin dose levels. One explanation could be that the relationship observed between zalfermin and glucose tolerance may be influenced by body weight; however, further investigation is needed to assess this.
Some FGF21 analogs have shown sustained effects on insulin sensitivity and trends in the lowering of FPG [13,24]. However, in two previous studies, the results observed were in populations with type 2 diabetes, and therefore, the effects on glycemic parameters may be more pronounced than in the current study [13-24]. Nevertheless, further studies are warranted to determine potential effects of zalfermin on glucose metabolism parameters in participants with types 2 diabetes.
This study had several limitations. Although the purpose of the trial was to primarily assess safety and tolerability to establish a well- tolerated dose range for zalfermin to be investigated in larger trials, the relatively small sample size of 57 participants, and short duration and follow-up time, may affect the generalizability of the safety and efficacy assessments. Furthermore, most of the population was White and no Asian participants were included, thus there was limited diversity in this trial. In addition, fewer females were included in the 27 and 60 mg treatment groups, thus, there are limited data on the effects of zalfermin on the menstrual cycle and reproductive organs at higher doses. Longer, larger clinical studies may provide further insights into the efficacy and safety of zalfermin in more diverse populations.
Based on the safety and PK profile established during this MAD study of subcutaneous zalfermin, the maximum tolerated dose was 60 mg, and this was compatible with a once-weekly dosing regimen. The PD profile of zalfermin, particularly the improvement in the plasma lipid profile, is promising for the treatment of a range of cardiometabolic diseases such as MASH and dyslipidemia. The results obtained from this trial support further clinical development of zalfermin.
Acknowledgments
The authors thank the trial participants, the investigators, and trial site staff who conducted the trial. Medical writing support was provided by Casey McKeown, RVN, FdSc, and Liam Gillies, PhD, of Titan, OPEN Health Communications, and funded by Novo Nordisk, in accordance with Good Publication Practice (GPP) guidelines (www.ismpp.org/gpp-2022).
Data availability Access request proposals can be found at novonordisk-trials.com
Funding
This study was funded by Novo Nordisk A/S.
Conflict of interest disclosure
K.D., J.S.H., M.S.P., S.L.L., O.B., S.T., and B.A. are all employees of and shareholders in Novo Nordisk. C.K. is an employee of and shareholder in ICON.
Ethics approval
The trial was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. The protocol for this trial was approved by an independent ethics committee/institutional review board.
Participant consent
All participants provided written informed consent.
Clinical trial registration
This study is registered with ClinicalTrials.gov (NCT03479892).
Author contributions
K.D., J.S.H., M.S.P., S.L.L., O.B., S.T., and B.A. wrote the manuscript; K.D., J.S.H., M.S.P., S.T., and B.A. designed the research for the SAD and MAD studies; K.D., J.S.H., S.T., and B.A. performed the research for the SAD and MAD studies; K.D., J.S.H., M.S.P., S.T., and B.A. analyzed the data; S.L.L. and O.B. contributed new reagents and analytical tools; C.K. was the principal investigator and responsible for assessment of adverse events as well as the medical care of the participants during the study.
References
- Rinella ME, Lazarus JV, Ratziu V, et (2023) A multisociety Delphi consensus statement on new fatty liver disease nomenclature. J Hepatol. [crossref]
- Takahashi Y, Fukusato T (2014) Histopathology of nonalcoholic fatty liver disease/ nonalcoholic World J Gastroenterol. [crossref]
- Chalasani N, Younossi Z, Lavine JE, et al. (2018) The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Hepatology. [crossref]
- Murag S, Ahmed A, Kim D (2021) Recent epidemiology of nonalcoholic fatty liver Gut Liver. [crossref]
- Younossi ZM, Koenig AB, Abdelatif D, et al. (2016) Global epidemiology of nonalcoholic fatty liver disease:Meta-analytic assessment of prevalence, incidence, and Hepatology. [crossref]
- Le MH, Yeo YH, Li X, et (2022) 2019 global NAFLD prevalence: a systematic review and meta-analysis. Clin Gastroenterol Hepatol. [crossref]
- Harrison SA, Bedossa P, Guy CD, et (2024) A phase 3, randomized, controlled trial of resmetirom in NASH with liver fibrosis. N Engl J Med. [crossref]
- Kharitonenkov A, Shiyanova TL, Koester A, et (2005) FGF-21 as a novel metabolic regulator. J Clin Invest. [crossref]
- Xu J, Lloyd DJ, Hale C, et (2009) Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet- induced obese mice. Diabetes. [crossref]
- Xie T, Leung PS (2017) Fibroblast growth factor 21: a regulator of metabolic disease and health span. Am J Physiol Endocrinol Metab. [crossref]
- Keinicke H, Sun G, Mentzel CMJ, et al. (2020) FGF21 regulates hepatic metabolic pathways to improve steatosis and Endocr Connect. [crossref]
- Kliewer SA, Mangelsdorf DJ (2019) A dozen years of discovery: insights into the physiology and pharmacology of FGF21. Cell Metab. [crossref]
- Kaufman A, Abuqayyas L, Denney WS, et al. (2020) AKR-001, an Fc-FGF21 analog, showed sustained pharmacodynamic effects on insulin sensitivity and lipid metabolism in type 2 diabetes Cell Rep Med. [crossref]
- Rosenstock M, Tseng L, Pierce A, et al. (2023) The novel glycoPEGylated FGF21 analog pegozafermin activates human FGF receptors and improves metabolic and liver outcomes in diabetic monkeys and healthy human volunteers. J Pharmacol Exp Ther. [crossref]
- Sass-Ørum K, Tagmose TM, Olsen J, et al. (2024) Development of zalfermin, a long- acting proteolytically stabilized FGF21 analog. J Med Chem. [crossref]
- Dahl K, Hansen JS, Clausen J, et al. (2022) SAT116 – Sustained reduction of triglyceride and LDL cholesterol from single administration of the novel long-acting FGF21 analogue 0499. J Hepatol.
- Ahart ER, Phillips E, Wolfe MS, Marsh C (2021) The role of kisspeptin in the ovarian cycle, pregnancy, and IntechOpen.
- Zhang Y, Cao X, Chen L, et al. (2020) Exposure of female mice to perfluorooctanoic acid suppresses hypothalamic kisspeptin-reproductive endocrine system through enhanced hepatic fibroblast growth factor 21 synthesis, leading to ovulation failure and prolonged J Neuroendocrinol. [crossref]
- Singhal G, Douris N, Fish AJ, et (2016) Fibroblast growth factor 21 has no direct role in regulating fertility in female mice. Mol Metab. [crossref]
- Owen BM, Bookout AL, Ding X, et al. (2013) FGF21 contributes to neuroendocrine control of female Nat Med. [crossref]
- S. Food and Drug Administration (2018) Guidance for industry: Bioanalytical method validation.
- European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP) (2011) Guideline on bioanalytical method validation.
- Dong JQ, Rossulek M, Somayaji VR, et al. (2015) Pharmacokinetics and pharmacodynamics of PF-05231023, a novel long-acting FGF21 mimetic, in a first- in-human Br J Clin Pharmacol. [crossref]
- Gaich G, Chien JY, Fu H, et al. (2013) The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab. [crossref]
- Loomba R, Lawitz EJ, Frias JP, et al. (2023) Safety, pharmacokinetics, and pharmacodynamics of pegozafermin in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 1b/2a multiple-ascending-dose Lancet Gastroenterol Hepatol. [crossref]
- Alkhouri N (2022) Pharmacokinetic (PK) and pharmacodynamics (PD) of BIO89- 100, a novel glycoPEGylated FGF21, in non-alcoholic steatohepatitis (NASH) patients with compensated J Hepatol.
- Kim AM, Somayaji VR, Dong JQ, et al. (2017) Once-weekly administration of a long-acting fibroblast growth factor 21 analogue modulates lipids, bone turnover markers, blood pressure and body weight differently in obese people with hypertriglyceridaemia and in non-human primates. Diabetes Obes Metab. [crossref]
- Charles ED, Neuschwander-Tetri BA, Frias JP, et (2019) Pegbelfermin (BMS- 986036), PEGylated FGF21, in patients with obesity and type 2 diabetes: results from a randomized phase 2 study. Obesity (Silver Spring) [crossref]
- Talukdar S, Owen BM, Song P, et (2016) FGF21 regulates sweet and alcohol preference. Cell Metab. [crossref]
- von Holstein-Rathlou S, BonDurant LD, Peltekian L, et al. (2016) FGF21 mediates endocrine control of simple sugar intake and sweet taste preference by the Cell Metab. [crossref]
- Loomba R, Sanyal AJ, Kowdley KV, et al. (2023) Randomized, controlled trial of the FGF21 analogue pegozafermin in NASH. N Engl J Med. [crossref]
- Harrison SA, Frias JP, Neff G, et (2023) Safety and efficacy of once-weekly efruxifermin versus placebo in non-alcoholic steatohepatitis (HARMONY): a multicentre, randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Gastroenterol Hepatol. [crossref]
- gov (2005) Research study on whether a combination of 2 medicines (NNC0194 0499 and semaglutide) works in people with non-alcoholic steatohepatitis (NASH)
- Lau DCW, Batterham RL, le Roux CW (2022) Pharmacological profile of once- weekly injectable semaglutide for chronic weight Expert Rev Clin Pharmacol. [crossref]
- Antonellis PJ, Kharitonenkov A, Adams AC (2014) Physiology and Endocrinology Symposium: FGF21: insights into mechanism of action from preclinical studies. J Anim Sci. [crossref]
- Bhatt DL, Bays HE, Miller M, et al. (2023) Pegozafermin for the treatment of severe hypertriglyceridemia: a randomized, double-blind, placebo-controlled phase 2 study (ENTRIGUE STUDY) Metab Clin Exp.