Summary
Macrophage Activation Syndrome (MAS) is a disorder related to hemophagocytic lymphohistiocytosis and is a life-threatening complication of rheumatic diseases. The diagnosis is challenging because MAS symptoms are quite similar to those of many active autoimmune diseases or severe sepsis. We describe the case of a female patient with systemic lupus erythematosus that presented with symptoms suggesting acute decompensation of autoimmune disease and sepsis. She was later diagnosed with MAS. Despite an aggressive immunosuppressive treatment, she developed a fatal outcome.
Keywords
Macrophage activation syndrome, Systemic lupus erythematosus, Hemophagocytic lymphohistiocytosis, Infection
Introduction
Macrophage Activation Syndrome (MAS) is a disorder related to hemophagocytic lymphohistiocytosis (HLH) and is a life-threatening complication of rheumatic diseases [1]. HLH is divided into primary and secondary. While primary (or familial) HLH is an inherited disease, secondary HLH is triggered by other diseases, including infections, malignancy, and autoimmune diseases [2]. MAS is a secondary HLH associated with autoimmune diseases. The most common is Systemic Juvenile Idiopathic Arthritis (SJIA), followed by Systemic Lupus Erythematosus (SLE) [3].
The diagnosis is challenging because MAS symptoms are quite similar to those of many active autoimmune diseases or severe sepsis. The typical signs and symptoms are persistent fever, hepatosplenomegaly, lymphadenopathy, and hemorrhagic manifestations. Abnormal results include cytopenia, coagulopathy, and hyperferritinemia. Since the mortality rate is up to 50% in adults, early recognition of MAS is important to improve prognosis [4].
We describe a case report of a SLE female patient that presented with symptoms suggesting acute decompensation of the auto-immune disease and sepsis. She was later diagnosed with MAS. Despite aggressive immunosuppressive treatment, she developed a fatal outcome.
Case Report
A 29-year-old female patient with a known diagnosis of SLE since 2013 was admitted to our Department with a 38.5°C fever, diffuse edema, and dyspnea with one-week duration. Her medical history was characterized by severe SLE with recent exacerbation of the disease while on maintenance treatment with azathioprine and oral low-dose corticosteroids. Therefore, she was started on treatment with cyclophosphamide. At admission, laboratory examination revealed anemia (hemoglobin of 4.3 mg/dL), leucocytes of 8,980/mm3 (35% immature forms), and low platelet count (83,000 per microliter of blood). There were signs of renal impairment function (creatinine of 1.97 mg/dl) and proteinuria (urinary protein-to-creatinine ratio of 1.8). The albumin concentration was low (0.8 g/dL). The complement levels were reduced (C3 of 70 mg/dL and C4 of 30 mg/dL). An abdominal ultrasound revealed splenomegaly.
At this point, cyclophosphamide was withdrawn due to the possibility of medication toxicity. She was kept on a high dose of prednisone. Given the possibility of systemic infection, the patient was started on large-spectrum antibiotics (piperacillin plus tazobactam). An upper Gastrointestinal (GI) endoscopy revealed signs of esophageal candidiasis, but no ulcer or gastritis. Therefore, oral fluconazole was also started. Blood and urine cultures posteriorly showed negative results.
Despite the treatment with antibiotics and antifungals and the suspension of cyclophosphamide, the patient presented an unfavorable evolution. After 3 days of blood and filtered platelet transfusion, she recurred with anemia (hemoglobin of 5.2 mg/dL) and low platelets (13,000 per microliter of blood). Additional blood exams also revealed leucopenia (5,120/mm³), normal fibrinogen (216 mg/dL), and high ferritin levels (10,508 ng/mL – reference of 13-150 ng/mL). The aspartate aminotransferase was elevated (65 units/L), as well as triglycerides (236 mg/dL). After a few days, the renal function continued to deteriorate (creatinine level of 2.86 mg/dL) with metabolic acidosis.
A bone marrow biopsy was performed after hematologic consultation, showing evidence of hemophagocytosis (Figure 1). At this point, treatment was switched to high dose of methylprednisolone (1000 mg per day) associated with intravenous immunoglobulin (30 g per day for 5 days). Despite this approach, on the third day, the patient presented with massive GI bleeding. A new endoscopy did not reveal any ulcer or severe gastritis. In a few days, respiratory failure treated with mechanical ventilation occurred, with the patient dying due to multiple organ failures.
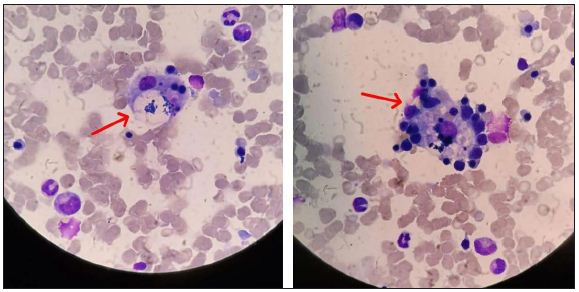
Figure 1: Bone marrow biopsy showing focal signs of hemophagocytosis (arrows). Hematoxylin and eosin stained, 1000× magnification.
Discussion
MAS is a secondary HLH related to rheumatic diseases, and it was first described as a complication of SJIA in 1985 by Hadchouel et al. [5]. It is a life-threatening condition characterized by cytopenia, high fever, liver insufficiency, andl coagulopathy.
Excessive activation and proliferation of T lymphocytes and macrophages or histiocytes lead to extensive hemophagocytosis in the bone marrow and cytokine storm [1]. The exact incidence of MAS in rheumatic diseases is still unknown. Although MAS is by far the most common disease in the pediatric population with SJIA, there have been increased reports of MAS related to SLE. The occurrence of MAS-associated SLE in adults is uncommon, ranging from 0.9 to 4.6%. It usually occurs in young female patients [3]. The clinical characteristics of MAS-associated SLE and active SLE are very similar. Both entities share clinical and laboratory features, which include fever, cytopenia, and splenomegaly. This makes the differential diagnosis very difficult. Hyperferritinemia is considered the best parameter to distinguish between MAS and SLE, with a sensitivity and specificity of about 100% favoring MAS [6]. Following multiple organ failures, if MAS is left untreated or even undetected, the mortality rate can rise to 42% in adults [4]. The index of suspicion for MAS is higher when an infection is ruled out or inflammation persists and does not respond to treatment of an underlying infection. In the present case, it could be useful to start an immunomodulatory therapy even in the face of infection. Systemic antibiotics were started as soon as the patient was admitted, even in the absence of a clear infection [7]. Although an identifiable precipitating factor is often not clearly identified, MAS has been related to numerous triggers, including among others a flare of the underlying disease, the toxicity of nonsteroidal antiinflammatory drugs, and viral infections [2]. Most cases are triggered by high activities of autoimmune diseases or infectious agents, resulting in a prolonged immune activation, predominantly by the cytotoxic T cells and the macrophages [3]. In a systematic review of the literature, Aziz et al. described the major risk factors that led to the development of MAS in SLE [8]. According to these authors, the most important precipitating factors were the lupus flare itself, its time of onset, and a high systemic lupus erythematosus disease activity index. Other factors identified were infections, drugs, underlying malignancy, and pregnancy [3,8]. Although there is no obvious cause of MAS occurrence in the present case, we considered the presence of systemic infection as the most probable cause. However, one should consider SLE decompensation or cyclophosphamide toxicity as potential triggers.
In 2014, Fardet et al. developed the HScore, a clinical tool that may be used to determine the probability of getting secondary HLH in adults [9]. This score encompasses 9 variables (known underlying immunosuppression, high temperature, organomegaly, triglyceride, ferritin, serum glutamic oxaloacetic transaminase, fibrinogen, cytopenia, and hemophagocitosys features on bone marrow aspirate). Each variable has a distinct weight, as reported by Fardet et al. [9] (Table 1).
Table 1: The HScore. Reproduced from Fardet et al. (2014) with permission of John Wiley and Sons.
|
Parameter |
No. of points (criteria for scoring) |
| Known underlying immunosuppression | 0 (no) or 18 (yes) |
| Temperature (°C) | 0 (<38.4)
33 (38.4-39.4) 49 (>39.4) |
| Organomegaly | 0 (no)
23 (hepatomegaly or splenomegaly) 38 (hepatomegaly and splenomegaly) |
| Number of cytopenias | 0 (one lineage)
24 (two lineages) 34 (three lineages) |
| Ferritin (ng/mL) | 0 (<2000)
35 (2000-6000) 50 (>6000) |
| Triglyceride (mmol/L) | 0 (<1.5)
44 (1.5-4) 64 (>4) |
| Fibrinogen (g/L) | 0 (>2.5)
30 (<2.5) |
| Aspartate Aminotransferase (U/L) | 0 (<30)
19 (>30) |
| Hemophagocytosis on bone marrow | 0 (no)
35 (yes) |
According to the authors, the probability of having hemophagocytic syndrome ranged from <1% with an HScore of ≤90 to >99% with an HScore of ≥250 [9]. In the present case, the HScore was 263, suggesting a greater than 99% probability of having secondary HLH/MAS.
To date, several therapeutic options are available, including non-biologic and biologic treatments. The mainstay of MAS treatment is glucocorticoid therapy, usually with intravenous methylprednisolone 30 mg/Kg/dose (maximum 1 g) for 1 to 3 days. For the nonresponders, additional therapy with cyclosporin is recommended. For patients who are refractory to the high dosages of the above medications, alternative options like etoposide, cyclophosphamide, and plasma exchange can be useful, although no randomized trials are showing consistent results of these medications [3]. Recently, reports with biological agents in refractory cases have shown promising results, including infliximab, rituximab, and intravenous immunoglobulin [7].
In the present case, some factors may have contributed to the unfavorable outcome. Firstly, we must acknowledge that there was some delay in the proper diagnosis of MAS. Secondly, despite the recent use of cyclophosphamide and oral corticosteroid, none of these therapies were the first-line treatment for MAS; therefore, the institution of high-dose methylprednisolone contributed to an additional delay in correct treatment. Finally, our patient was treated in a public tertiary Brazilian institution that lacks the more recent options for the treatment of MAS refractory cases, like immunobiological agents. Consequently, we could only opt for intravenous immunoglobulin in the present case.
Conclusions
MAS is a challenging and life-threatening disorder related to HLH. The occurrence of MAS-associated SLE in adults is relatively uncommon. The clinical characteristics of MAS-associated SLE and active SLE are very similar. Both entities share clinical and laboratory features, which include fever, cytopenia, and splenomegaly. This makes the diagnosis very difficult. A high index of suspicion, associated with immediate treatment is essential for the achievement of better outcomes.
References
- Dhote R, Simon J, Papo T, Detournay B, Sailler L, et al. (2003) Reactive hemophagocytic syndrome in adult systemic disease: report of twenty-six cases and literature review. Arthritis Rheum 49: 633-639. [crossref]
- Ravelli A (2002) Macrophage activation syndrome. Curr Opin Rheumatol 14: 548-552.
- Lerkvaleekul B, Vilaiyuk S (2018) Macrophage activation syndrome: early diagnosis is Open Access Rheumatol 10: 117-128. [crossref]
- Crayne CB, Albeituni S, Nichols KE, Cron RQ (2019) The immunology of macrophage activation Front Immunol 10: 119. [crossref]
- Hadchouel M, Prieur AM, Griscelli C (1985) Acute hemorrhagic, hepatic, and neurologic manifestations in juvenile rheumatoid arthritis: possible relationship to drugs or infection. J Pediatr 106: 561-566. [crossref]
- Egües Dubuc CA, Uriarte Ecenarro M, Meneses Villalba C, Aldasoro Cáceres V, Hernando Rubio I, et al. (2014) Hemophagocytic syndrome as the initial manifestation of systemic lupus Reumatol Clin 10: 321-324. [crossref]
- Granata G, Didona D, Stifano G, Feola A, Granata M (2015) Macrophage activation syndrome as onset of systemic lupus erythematosus: a case report and a review of the Case Rep Med 2015: 294041. [crossref]
- Aziz A, Castaneda EE, Ahmad N, Veerapalli H, Rockferry AG, et al. (2021) Exploring macrophage activation syndrome secondary to systemic lupus erythematosus in adults: a systematic review of the literature. Cureus 13: e18822. [crossref]
- Fardet L, Galicier L, Lambotte O, Marzac C, Aumont C, et (2014) Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol 66: 2613-2620. [crossref]