DOI: 10.31038/CST.2019424
Summary
Cytokines are molecular messengers that mediate cell-to-cell communication in an autocrine and paracrine fashion. Development of cytokine-based drugs requires a deep understanding of cytokine biology and continuous engineering effort to fine-tune pharmacological properties to elicit potent anti-tumor response while keeping toxicity to a minimum. To date, high dose bolus IL-2 for metastatic melanoma and INF-α for renal carcinoma have been approved as single agents for cancer therapy. In additions, immune cytokines, especially IL-2 and more recently IL-7, IL-15, and IL-21, have been used in Adoptive T-Cell Therapy (ACT) to expand anti-tumor T cells. Nonetheless, systemic monotherapy using cytokines has not fulfilled the promise of clinical efficacy due to several limitations, including failure to achieve effective concentrations in the tumor tissue, severe systemic toxicities that often associated with the high dose administration, and unwanted induction of immune suppression. To address these impediments, innovative approaches have been adopted to improve cytokine-based immunotherapy. These include PEGylation, structure-based cytokine engineering and antibody-cytokine fusion molecules. In this review, we summarize the clinical activity of immune cytokines in the treatment of cancers and recent progress in the engineering of new cytokine therapeutics.
1. Introduction
The role of cytokines in cancer immunology
Immune cells are well recognized to play critical functions in the surveillance of tumor cells and the eradication of established disease foci. A weakened or exhausted immune system usually leads to the growth of tumor cells and metastases. Current immune oncological therapies, such as the immune checkpoint blockade agents PD-1/PD-L1 antibodies, and CAR T therapy, are examples of either boosting cytotoxic T cell functions or directly providing tumor specific T cells to combat tumors.
Immune cytokines are small soluble proteins that function to maintain the activity of the immune system by playing roles in many aspects, such as the proliferation and differentiation of cytotoxic T cells and enhance effector cell antitumor cytotoxic activities. Ever since the identification of the pro-inflammation activity of Interleukin-2 (IL-2), immune cytokines have been tested as cancer therapeutic. However, due to their broad and systemic effect, few cytokines could clinically demonstrate convincing anti-tumor activity with limited adverse effects. Nevertheless, clinical trials with native or engineered cytokines provide valuable insights on how immune networking is established and will shed light on future tumor immune therapy.
2. Current Clinical Experience with Immune Cytokines in Cancer Therapy
2.1 IL-2
2.1.1 Native or Recombinant IL-2
Interleukin-2 (IL-2) is a cytokine produced mainly by activated CD4 T cells and CD8 T cells, and occasionally by certain B cells and dendritic cells [1]. IL-2 can stimulate the proliferation of T cells and other cells that express IL-2 receptors. Since cytotoxic T cells play roles in tumor immunity by attacking cancerous cells directly, IL-2 was used as an agent to boost cytotoxic T cell function to treat cancer patients.
In fact, IL-2 was one of the first immune cytokines tested in clinical trials as early as the 1980s. Initially, only purified native IL-2 was available, and due to the limitation of the purification process, only small doses of IL-2 were used. Unfortunately, only toxicity but no clinical anti-tumor activity was observed when tested in advanced cancer patients [2].
Later, when recombinant IL-2 (rIL-2) was produced, higher doses of IL-2 were tested together with autologous LAK cells in 25 patients with metastatic cancer, who had failed standard therapy. In this trial, very promising anti-tumor activity was observed [3]. One 33-year old female with metastatic melanoma was among the first cohort of patients who received the high dose rIL-2 treatment. This patient responded very well to this treatment, with complete response observed in a few months. In addition, she was cancer-free for at least 29 years.
Additional clinical trials revealed that high dose rIL-2 alone was sufficient to obtain therapeutic activity that leads to tumor regression. In one clinical trial, high dose bolus IL-2 treatment led to a 15% objective response rate (ORR) in 182 metastatic melanoma patients and a 19% ORR in 277 metastatic renal cancer patients. After several multi-institutional studies demonstrated the clinical efficacy of rIL-2 treatment, FDA approved rIL-2 to treat metastatic renal cancer and melanoma in 1992 and 1998, respectively.
Due to the short half life of IL-2, which is about 7 minutes in humans, IL-2 has to be administered at a large dose and several times a day. This limits the use of IL-2 as an effective therapeutic agent. Side effects include hypotension, heart toxicities, and vascular leak syndrome. These side effects prevent many patients from taking rIL-2 at the optimal high dosage. Initially, treatment related death happened in 2~4% of IL-2 treated patients, but the mortality rate was reduced to less than 1% later with appropriate clinical management [4].
In addition to serving as a direct therapeutic drug to cancer patients, IL-2 is also used in cancer immune therapies. Both CAR-T and ACT therapies are dependent on the availability of a large amount of T cells, either engineered T cells or endogenous antitumor lymphocytes. Thus, IL-2 has been used as the growth factor to expand these types of cells in vitro to obtain enough cells to treat patients.
2.1.2 Engineered IL-2
NKTR-214:
The IL2 receptor complex has three distinct subunits, the α-chain (IL-2Rα or CD25), β-chain (IL-2Rβ or CD122), and γ-chain (γc or CD132). Not all 3 receptors exist on all types of IL-2 target cells. IL-2Rα and L-2Rβ are commonly found as heteroreceptors in many target cells, but the presence of the α-chain in the receptor complex increases the affinity for IL-2 by 2 orders of magnitude [5]. Treg cells, the immune suppressive T cells, express CD25 and thus have the highest affinity for IL-2 as compared to other T cells. This explains why at a lower dose, IL-2 failed to show any anti-tumor activity in clinical trials. To limit IL-2Rα binding, a PEGylated form of rIL-2, which is named NKTR-214, has been developed.
PEGylation is a common practice to extend the in vivo half-life of peptides or small proteins by covalent or non-covalent attachment of polyethylene glycol (PEG) to peptides/proteins. For NKTR-214, IL-2 was conjugated with releasable PEG chains at lysine residues in the IL-2Rα-binding interface. Since the bulky PEG chains block the interaction of IL-2 with its receptor, NKTR-214 serves as a prodrug, and after several PEG chains are released, the one-chain pegylated IL-2 (1-PEG-IL-2) becomes the active form. Compared with IL-2, 1-PEG-IL-2 has comparable binding affinity to IL-2Rβ but much less affinity to IL-2Rα, and thus has a drastically lowered affinity to IL-2Rαβγ, the receptor complex that is abundant on Treg [6].
As expected, NKTR-214 increases the half-life of IL-2 to 15.5 hours in mice. Since the active form is slowly released, the Cmax is reached 24 hrs after a single treatment. More importantly, compared to IL-2, the active forms of NKTR-214 have a 10-fold lower Cmax, but 27-fold higher AUC, meaning a controlled release and more drug exposure as the benefit of PEGylation [6].
In the mouse B16F10 melanoma model, NKTR-214 demonstrated better activity and safety than rIL-2. While rIL-2 had to be administered twice daily, NKTR-214 was given to mice every 9 days. Single agent NKTR-214 treatment had much better anti-tumor activity than that of IL-2 [7].
Analysis of T cell populations in mice treated with NKTR-214 also indicated that total and memory CD8 T-cell population in tumor infiltrating lymphocytes were significantly increased. This is more sustainable and stable than the effect of natural IL-2.
When combined with the immune checkpoint inhibitor CTLA-4 antibody, NKTR-214 demonstrated synergistic activity to control tumor growth in the EMT6 breast cancer model, with 70% of mice showing complete response and becoming tumor free. In comparison, the tumor free rate in mice treated with IL2 and anti-CTLA4 is 40%. Tumor free mice were resistant to tumor- rechallenges, indicating a durable and specific immunity had been established with the combination of NKTR-214 and checkpoint blockade [7].
Due to the slow release of the active cytokines, longer half life and reduced peak concentration, NKTR-214 also demonstrated less severe side effects in animal studies. rIL-2 treated mice became hypothermic and exhibited shivering behavior, while NKTR-214 treated mice only showed mild weakness [7] .
Although NKTR-214 was designed to spare Treg, in vivo studies revealed that at the high dosage used in tumor treatment, activation of Treg may not even be an issue for rIL-2. To the opposite, the Treg population was reduced by both IL-2 and NKTR-214 as compared with control treatment.
Using a humanized model of IL-2 therapy, Li and colleagues reported that Treg in fact controls the toxicity during IL-2 therapy [8]. Down-regulation of Treg by high dose rIL-2 or NKTR-214 thus could be directly linked to the toxicity related to IL-2 therapies. To ameliorate IL-2 toxicity, a strategy is to use the PIM-1 kinase inhibitor, Kaempferol [8]. As discovered previously, Kaempferol enhances the suppressive function of Treg cells by inhibiting FOXP3 phosphorylation by PIM1[9, 10].
With the promising activity in animal models, NKTR-214 has been tested in clinical trials. However, when NKTR-214 was tested as monotherapy in the early EXCEL trial, no objective response was observed, compared with the 15~29% ORR for rIL-2 seen in other trials. The lack of clinical efficacy for NKTR-214 monotherapy was accounted for by a lower maximum-administered dose (0.012 mg/kg q3w), the much lower Cmax and weaker activation of lymphocytes. Analysis of clinical trial data also revealed that NKTR-214 has a mixed effect on Tregs. While tumor-infiltrating Treg cells are reduced, peripheral Treg populations are increased after NKTR-214 treatment.
When tested in combination as first line treatment in early stage clinical trials, NKTR-214 and PD-1 antibody Nivolumab produced 64% ORR in stage IV melanoma and 71% ORR in Stage IV RCC. The same combination also produced 60% ORR as first or second line treatment in Stage IV NSCLC [11]. Again, additional trials are needed to verify the clinical efficacy for the combination strategy and to show its benefit over Nivolumab monotherapy.
In terms of side effects, NKTR-214 has not solved all of the adverse effects that IL-2 displays. Still, many patients report having fatigue, rashes, and flu-like symptoms.
Neo-2/15
Instead of using pegylation to block binding to IL-2Rα receptor, a creative de novo approach was taken to develop IL-2 mimics with better selectivity for IL-2Rβγ and higher in vivo stability [12]. Although called “de novo” design, this strategy started with IL-2 as the template, extracting α helices that interacting with IL-2Rβγ, but modified the loops connecting the helices and the backbones to obtain structures with lower energy. Site mutations were then introduced into the lead structure for a better affinity to IL-2Rβγ.
The final structure, Neo-2/15, shows only 14% and 24% structure-based sequence identity to human and mouse IL-2 respectively, but binds to IL-2Rβγ receptor of both species with very high affinity. As designed, Neo-2/15 has no detectable binding to IL-2Rα and demonstrates thermal stability at 80°C[12].
As demonstrated by in vivo studies, Neo-2/15 causes less expansion of the immunosuppressive Treg cells than IL-2. When mice were treated with Neo-2/15, the ratio of CD8 T cells over Treg cells was greatly increased. Most importantly, Neo-2/15 has been found to have a superior immunotherapeutic activity to IL-2 in mouse models of melanoma and colon cancer. When Neo-2/15 was used in combination with TA99, which is an antibody against TRP-1, 4 out of 10 mice were tumor-free after treatment. Neo-2/15 caused less weight loss and general health problems in treated mice as compared with native IL-2 [12].
In addition to NKTR-214 and Neo2/15, there are several other engineered IL-2 cytokines under clinical testing. An IL-2 variant (IL2v), which has abolished binding to IL2Rα, was recombinantly linked to anti-FAP or anti-CEA antibodies [13]. The antibody- cytokine fusions (FAP-IL2v and CEA-IL2v) are designed to enrich cytotoxic T cells- specific IL-2 to tumor microenvironment and thus limiting its effect on peripheral Treg. Furthermore, CEA-IL2v and FAP-IL2v enhanced the cytotoxic activity of Natural Killer (NK) cells when combined with ADCC-competent antibodies against HER2 or EGFR [13].
2.2 IL-10
T cell exhaustion is frequently observed in cancer patients. It could lead to the escape of tumor cells from immune surveillance and create hurdles for successful cancer immunotherapy.
As one of the immune cytokines having dual roles, IL-10 is anti-inflammatory at low concentrations, but pro-inflammatory at high concentrations to induce immune activation and invigorate CD8+ T cells. It is produced by activated T cells, monocytes and lymphocytes, which is further induced by PD-1 blockade in animal studies [14]. It is proposed that PD-1 induces IL-10 secretion and renders tumor microenvironment immunosuppressive, as evidenced by the synergistic anti-tumor effect by anti-PD-1 and anti-IL-10 antibodies on ovarian cancer xenografts.
Nevertheless, when PEGylated IL-10 (also known as Pegilodecakin, or AM0010) was administered to mice to obtain sustained elevated serum concentration, activation of tumor-resident CD8+ T cells was observed and tumor rejection occurred [15, 16].
A phase I clinical trial was conducted in patients with intermediate to poor-risk renal cell cancer. In patients treated with the optimal dose of AM0100, which was 20 μg/kg, partial responses were observed in four out of 15 patients, resulting in an overall response rate of 27%. Furthermore, a prolonged stable disease of at least 4 months was observed in 4 patients, with one having disease stabilization for 20 months. [17]
In this trial, patients self-administered PEGylated IL-10 subcutaneously at doses of 1 to 40 μg/kg once daily [18]. At 29 days after treatment initiated, sustained serum levels of IFNγ, IL-18, IL-4, and IL-7 were detected. Patients with better clinical response tended to have increased LAG+PD1+CD8+ T cells after the treatment, suggesting an invigoration of exhausted T cells after PEGylated IL-10 treatment. Patients with PR also have more T cell clones amplified. The combination of PEGylated IL-10 and the anti-PD1 antibody pembrolizumab was also tested in heavily pre-treated cancer patients, including melanoma, non-squamous cell lung cancer or RCC. 42% ORR was observed in 19 evaluable patients [18].
Observed adverse events (AEs) included anemia, fatigue, thrombocytopenia, fever, and injection site reactions. Grade 3 to 4 nonhematopoietic treatment-related AEs was observed in 5 out of 33 patients, Grade 3 to 4 anemia or thrombocytopenia was also observed in five patients. However, most treatment-related AEs were transient or reversible, and AM0100 was considered to have an acceptable toxicity profile. Prolonged exposure to AM0100 did not lead to acute toxicities at the therapeutic dose [17].
2.3 IL-12
The IL-12 family cytokines comprise heterodimeric IL-12, IL-23, IL-27, and IL-35. Although similar in structures, these cytokines have various biological and immunological functions. IL-12 is a pro-inflammatory cytokine produced by DCs, macrophages and B cells. Secretion of IL-12 can be potentiated by IFN-γ from T cells. IL-12 can further act on NK cells to induce IFN-γ [19].
Like other members of the IL-12 family, IL-12 is composed of 2 subunits: α and β. IL-12 shares the α subunit (p35) with IL-35, and the β subunit (p40) with IL-23. The IL-12 receptor also includes two subunits: IL-12Rβ1 and IL-12Rβ2. Like the ligands, the IL-12 family receptors also share subunits. IL-12Rβ1 exists in the receptors for IL-12 and IL-23, and IL-12Rβ2 exists in the receptors for IL-12 and IL-35.
In preclinical studies, IL-12 is able to enhance anti-tumor activity and totally eradicate large established tumors [20], which is dependent on CD8 T cells and IFN-γ [21]. IL-12 is also critical to anti-tumor T cell immunity induced by the anti-PD-1 antibody, as eliminating IL-12 with anti-IL-12 antibody entirely wiped out the anti-tumor effect of anti-PD-1 antibody in an MC38 tumor model [22]. It is reported that the PD-1 antibody acts on CD8+ T cells to produce IFN-γ, which subsequently induces DC to release IL-12.
In clinical trials, as recombinant IL-12 has to be used at high doses due to its short half-life, significant and unacceptable toxicity sometimes occurs.
2.3.1 Early Clinical Trials with IL-12
Cutaneous T Cell Lymphoma (CTCLs): CTCLs are a family of non-Hodgkin lymphomas. It has 2 common forms: mycosis fungoides (MF) and Sezary syndrome (SS). Patients with CTCLs usually have defects in IL-12 and are associated with deficient IFN-γ production and depressed Th1 cells functions. In a phase I clinical trial with 10 CTCL patients, subcutaneous treatment with IL-12 resulted in 2 complete responses in 5 MF patients with extensive plaque. Only mild and short-lived adverse effects were noticed. In these patients, an increased proportion of cytotoxic CD8+ T cell was observed. In total, IL-12 treatment achieved an ORR of 56% (5 out of 9 patients) [23].
Hodgkin’s and non-Hodgkin’s Lymphoma: Younes et al reported a phase II study on the effect of IL-12 on HL (Hodgkin’s lymphoma) and NHL (non-Hodgkin’s B cell lymphoma) [24]. Objective responses were only observed in NHL patients. It was concluded that i.v. treatment was more effective than s.c. injections (40% v.s. 7%), and responses were better in follicular grade I/II lymphoma v.s. diffuse large B-Cell lymphoma. Patients with less severe diseases tended to have a better response. In a phase I clinical trial [25], recurrent NHL patients showed 69% ORR when treated with both rituximab and IL-12. However, a follow-up phase II study showed that the efficacy in the combination treatment was entirely due to rituximab.
Kaposi Sarcoma (KS): KS is a tumor localized in the skin, caused by HHV-8 (human herpesvirus 8). It is often associated with iatrogenic immunosuppression or HIV-related immunodeficiency. In a dose-escalating clinical trial with 24 patients [26], high dose IL-12 showed 71% ORR, and one patient showed complete tumor regression after continued IL-12 therapy for almost 5 years. Again, some patients experienced psychoneurological problems. IL-12 was also studied in the phase II study in combination with pegylated liposomal doxorubicin [27]. Impressively, ORR of this trial was 83%, with 25% treated patients showing complete response.
In general, as we extrapolated from clinical trials, high doses of IL-12 commonly leads to severe hematologic toxicity such as neutropenia, thrombocytopenia, hyperbilirubinemia, and hypoalbuminemia. While pre-treatment with a priming dose of IL-12 allowing administration of higher doses, this strategy shows no improvement regarding the therapeutic outcomes. In addition, consecutive injections of IL-12 will lead to adaptive responses and the significant decline of IFN-γ induction. It is unlikely that IL-12 will become an effective single agent treatment for cancer patients.
2.3.2 IL-12-based immunocytokines (Antibody-cytokine fusion proteins)
In an effort to improve therapeutic efficacy and safety, IL-12 has been explored as fusion proteins with tumor targeting antibodies. By accumulating in the tumor tissue or even directly killing cancer cells, the antibody-cytokine fusion proteins are expected to induce a strong inflammatory signal to enhance antitumor response.
One of these fusion proteins is AS1409, which contains a humanized antibody BC1 that binds to the ED-B variant of fibronectin and directs IL-12 to tumor-associated vasculature. In a phase I trial with 11 melanoma and 2 renal cell carcinoma patients, AS1409 treatment induced elevated serum levels of IFN-γ and interferon-inducible protein-10 (IP-10) in all patients, indicating activation of cell-mediated immune response. One melanoma patient had a partial response, while stable disease was seen in 5 other patients [28]. At the maximum tolerated dose of 15 μg/kg, most drug-related adverse events were grade 2 or less, including pyrexia, fatigue, chills, headache, vomiting, and transient liver function abnormalities. Unfortunately, plasma half-life of this fusion antibody was only 22 hours, and antidrug antibody responses were seen in all patients [28]. The shorter than expected half-life and immunogenicity issue will somehow limit the clinical development of AS1409.
Another example is NHS-IL12, which is IL-12 fused with a tumor necrosis-targeting human IgG1 (an anti-histone antibody) [29]. NHS-IL12 has demonstrated anti-tumor activity in pre-clinical studies with murine and dog tumor models [29, 30]. Irradiation that induces the necrosis of tumor cells is shown to potentiate NHS-IL12 anti-tumor activity [31]. Combination of NHS-IL12 with anti-PD-L1 antibody achieves complete tumor regression in the EMT-6 mammary tumor model [32].
2.3.3 Delivery of IL-12 cDNA
Therapeutic IL-12 is usually delivered as recombinant or engineered IL-12 proteins. Alternatively, IL-12 was also explored as gene therapy by delivering viral or plasmid vectors to express IL-12 in the host. In clinical trials, RTS-hIL-12 plus veledimex were studied in recurrent or progressive glioblastoma multiforme adult patients.
IL-12 expressing vector can also be delivered to therapeutic cells directly. To help CAR T therapy, T cells were engineered to express IL-12 to enhance the durability and help change the tumor microenvironment to allow better tumor penetration. A MUC-16 targeting CAR T therapy has been developed for recurrent ovarian cancer and is currently tested in a phase I clinical trial [33].
2.4 IL-15
IL-15, a cytokine mainly produced by activated myeloid cells, shares structure similarity with IL-2 and binds to two of IL-2’s receptors: IL-2Rβ and γc. IL-15 has its own high affinity receptor, IL-15Rα. Due to receptor specificity, IL-15 acts on NK cells and activated T cells, but not on immunosuppressive Treg. For this reason, IL-15 is considered as an ideal cytokine to effectively augment CD8+ T cell and NK cell function in solid tumors.
Recombinant IL-15 produced in Escherichia coli was initially tested via intravenous bolus injection in advanced melanoma and RCC patients. Expansion of peripheral NK and CD8+ T cells was reported in patients treated with IL-15, but there was no objective clinical response [34]. rIL-15 was also tested subcutaneously in patients with a variety of solid tumors, and the best clinical outcome was stable disease [35]. In general, subcutaneously administration of rIL-15 tended to have a little higher dose-limiting toxicity at 3.0 μg/kg per day. The severe adverse events after patients were treated with rIL-15 included high fever, hypotension and thrombocytopenia [34].
To engineer a potent IL-15 ligand, IL-15 was recombinantly connected to the sushi domain of IL-15R alpha. The sushi domain functions as an IL-15 agonist by enhancing its binding and biological effects through the IL-2Rβ and γc heterodimer [36]. This super IL-15 ligand can be produced as an IgG- fusion protein to obtain longer half-life. ALT-803 is such an IgG fusion protein but contains a mutated IL-15 with superior activity [37]. In a mouse model of multiple myeloma, a single intravenous dose of ALT-803, but not IL15, eliminated well-established tumors and prolonged survival of mice. In B16F10 melanoma tumors and CT26 colon cancer models, ALT-803 also demonstrated better anti-tumor activity than IL-15, and increased peripheral blood lymphocyte, neutrophil, and monocyte counts by >8-fold [37].
In a phase one clinical trial in patients with hematologic malignancies who relapse after allogeneic hematopoietic cell transplantation (allo-HCT), ALT-803 was administered to 33 patients via the IV or subcutaneous (SQ) routes once weekly for 4 doses. In general, ALT-803 was well tolerated. While IV administration led to constitutional symptoms temporally related to increased serum IL-6 and IFN-γ, SQ delivery only resulted in prolonged (>96 hour) serum concentrations and only self-limited injection site rashes without acute constitutional symptoms. Activation of NK cells and CD8+ T cells, but not Treg, were observed after ALT-803 treatment. More importantly, clinical responses were observed in 19% of evaluable patients, including 1 complete remission lasting 7 months [38].
Nanoparticles and protein nanogels are potential efficient delivery methods for ALT-803. Nanogels contains an antibody molecule that can direct the delivery to a specific cell type, such as T cells. To receive the ALT-803 payload, T cells need to have a cell surface receptor as the “loading dock” that is not down-regulated after binding to the antibody. Tang et al identified CD45 acting as the stable, non-internalizing “loading dock” for nanogels [39]. After binding to the anti-CD45 antibody, CD45 remains to have close to 100% surface expression level for more than 60 hours. Once nanogels reach the surface of activated CD8+ T cells, the cytokine is released due to higher cell surface reduction potential.
Compared with free cytokines, nanogels delivery is able to selectively expanded T cells 16-fold in tumors. This also allows much higher doses of cytokine. In animal studies, ALT-803 nanogels are used to substantially enhance tumor eradication by mouse T cells or human CAR-T cells [39].
2.5 IFNα
Interferons (IFNs) play important roles in immune defense against cancer cells and intracellular pathogens. Based on their corresponding receptors, IFNs are categorized into 3 types: types I, II, and III. IFN-α belongs to type I IFN, which are generally expressed by all kinds of cells in response to viral infection. By promoting apoptosis in the infected cell, IFN-α interferes with viral replication. IFN-α also activates macrophages and NK cells.
IFN-α has been clinically used to treat a variety of malignancies, including indolent B cell lymphoma and hairy-cell leukemia, chronic myelogenous leukemia (CML), renal cell carcinoma (RCC), and melanoma.
In advanced RCC, IFN-α is approved for combination treatment with bevacizumab based on the results of the CALGB and AVOREN trials. In the CALGB trial, compared with IFN-α monotherapy, the addition of bevacizumab to IFN-α led to an improved ORR (25.5% vs. 13.1%, P < 0.0001) and a significant 3 month benefit in PFS (8.5 months vs. 5.2 months, P < 0.0001). However, overall survival (OS) did not significantly differ between the 2 groups (18.3 months vs. 17.4 months, unstratified log-rank, P = 0.097). In the AVOREN trial, median PFS was also significantly improved in the bevacizumab plus IFN-α arm at 10.2 months compared with 5.4 months in the IFN-α monotherapy group. Again, the final median OS was only marginally improved in combination arm (23.3 months vs. 21.3 months, HR = 0.91; 95% CI, 0.76–1.10, P = 0.34) [40].
The practical use of IFN-α in RCC management is limited, partially due to the frequency of IFN-α administration and the side effects of the treatment. IFN-α is administrated subcutaneously 3 times a week for 52 weeks. In the combination treatment group of the CALGB trial, 64% of patients need to reduce the dose due to intolerability, and 80% had severe toxicity (grade ≥3), including hypertension, anorexia, fatigue, and proteinuria. The majority of the patients (56%) discontinued for disease progression.
IFN-α is much less used now after anti-angiogenic tyrosine kinase inhibitors are approved for RCC. As a first line treatment of metastatic clear cell RCC, IFN-α is less effective than sunitinib, with much lower ORR (12% vs. 47%), mPSF (5.0 months vs. 11.0 months), and mOS (21.8 months vs. 26.4 months [41]. However, Naito et al reported that analysis of real world data suggested that IFN-α might have a better outcome in Japanese metastatic clear cell RCC patients than in the western patients [42].
In melanoma, IFN-α was used for more than a decade in the adjuvant setting, until the arrival of immune checkpoint inhibitors for adjuvant treatment of high-risk resected melanoma.
2.6 Interferon γ (IFN- γ)
IFN-γ, which is the only member in the type-II interferon family, is a dimerized soluble cytokine that is dominantly produced by natural killer (NK) cells, natural killer T (NKT) cells, CD4 helper T cells, and CD8 cytotoxic T cells. As an immune cytokine, IFN-γ’s function is not limited to promoting T cells / NK cells to migrate into the tumor tissue. It can also directly binds to its receptor on tumor cells and upregulates MHC class I antigen presentation. This enhances tumor recognition by cytotoxic lymphocytes, favoring tumor rejection.
In addition, IFN-γ also inhibits tumor angiogenesis by inducing the production of chemokines, such as MIG (CXCL9, a small cytokine belonging to the CXC chemokine family) and IP10 (CXCL10), through both direct mechanisms by reducing endothelial cell adhesion and indirect mechanisms by inducing the production of antiangiogenic molecules.
After dimerized IFN-γ binds to its receptor, which is composed of IFN-γ receptor 1 (IFNGR1) and IFN-γ receptor 2 (IFNGR2), it induces trans-phosphorylation and activation of JAK1 and JAK2. Activated JAK1/2 leads to the phosphorylation of IFN-γ receptor, providing STAT docking sites through the SH2 domain and to recruit STATs to the JAK- IFN-γ receptor complex. After recruited to the receptor complex, STATs become activated and form dimers, followed by translocation to the nucleus to promote the expression of target genes involved in cell proliferation, differentiation, and inflammation.
Because IFN-γ plays an important role in innate and adaptive immunity, loss of tumor sensitivity to IFN-γ will offer a mechanism for tumor cells to escape immune surveillance and to metastasize. This occurs when there is a defect in IFN-γ receptor or other molecules of the downstream signaling pathways, such as Jak1 and Jak2. A complete loss of tumor sensitivity to IFN-γR has been reported for human melanomas and lung adenocarcinomas, including the loss of IFN-γRα expression, loss of Jak1, and expression of an abnormally phosphorylated Jak2 enzyme [43]. Defects in IFN-γ signaling pathways also contribute to the resistance to immune checkpoint inhibitors in melanoma patients [44]. High expression of IFN-γ in melanoma patients is associated with longer overall survival after treatment with immune checkpoint inhibitor pembrolizumab [45].
Nevertheless, IFN-γ signaling can also pose a negative effect on cancer therapy. There are reports that IFN-γ down-regulates tumor antigen expression and promotes the emergence of tumor antigen-loss variants that leads to disease progression. A melanoma patient vaccinated with a gp100- derived class I peptide together with IFN-γ developed a metastatic lesion that was associated with a selective loss of gp100 expression after an initial response. This suggests that selective pressure facilitated the emergence of antigen-negative tumors.
IFN-γ is clinically used as an antiviral agent to treat infections. However, some patients under IFN-γ treatment are also cancer patients and thus provide insights on IFN-γ effects on cancers. Buddingh et al. reported a case of IFN-γ treatment of a 3-year old Acute Lymphoblastic Leukemia (ALL) patient [46]. In this case, IFN-γ was intended to restore immune function to treat severe systemic Candida dubliniensis infection due to chemotherapy-induced immunoparalysis. After the patient’s clinical condition was improved, imaging and abdominal ultrasound examination revealed that the patient was in remission, with complete resolution of brain lesions and greatly diminished kidney, liver and spleen lesions.
During the course of IFN-γ treatment, the percentage of HLA-DR-positive monocytes increased from almost 2% to 50%. The HLA-DR ratio is a good indication for innate immunity. Since this ALL patient had been treated with both chemotherapy and antifungal agents, IFN-γ was not the only reason for the complete control of fungal infection and long lasting remission. However, the contribution of IFN-γ to the normalization of immunity needs to be noted and worth further investigation.
AbZed
We have reported that IFN-γ is effective to enhance HER2-targeted antibody therapy. The anti-oncogenic HER2 (also know as p185erbB2/neu) antibody 7.16.4, when given at a sub-optimal dose of 1.5 mg/kg (1/3 of the normal dose), was unable to significantly inhibit the growth of the H2N113 tumor in syngeneic MMTV-neu transgenic mice. IFN-γ treatment alone also failed to cause significant inhibition of the growth of the tumor. However, the combination of low dose 7.16.4 and IFN-γ completely arrested the growth of H2N113 tumors [47].
A further step was taken to establish a fusion protein HER2 AbZed, which contains a scFv to bind to HER2, an Engineered Effector Domain (EED) to bind to the Fc region of IgG, and IFN-γ in a single fusion protein. Treatment with this engineered fusion protein is even more potent than the anti-HER2 antibody alone or simply combining scFv-ZZ and IFN-γ in terms of inhibiting tumor growth [48]. The new fusion protein demonstrates superior activity over the anti-HER2 antibody on tumors that are resistant to trastuzumab. Examination of tumor infiltrating macrophages and lymphocytes reveals that the fusion protein induces change in tumor microenvironments [48]. The EED domain is originally derived from the bacterial protein A but can be further humanized for therapeutic use.
3. Conclusion
Although some immune cytokines have been approved for the treatment of certain types of cancers, the clinical use is limited. PEGylation is able to extend the in vivo half-life of cytokines, but it remains unclear if such a modification can dramatically enhance the clinical utilities. While combination use of cytokines with other immunotherapeutic regimens appears to be the future, the systemic toxicity is one major limitation. This may be circumvented by recombinantly connecting a cytokine to target therapeutic agents, thus the cytokines can be selectively accumulated around tumor. It is hoped that by innovative engineering, immune cytokines may play a major role in immunotherapy against cancer.
Acknowledgement
We acknowledge grant supports from the Breast Cancer Research Foundation and the National Institutes of Health to M.I.G. (R01 CA219034). We thank Isabel Liang for her comments on the manuscript.
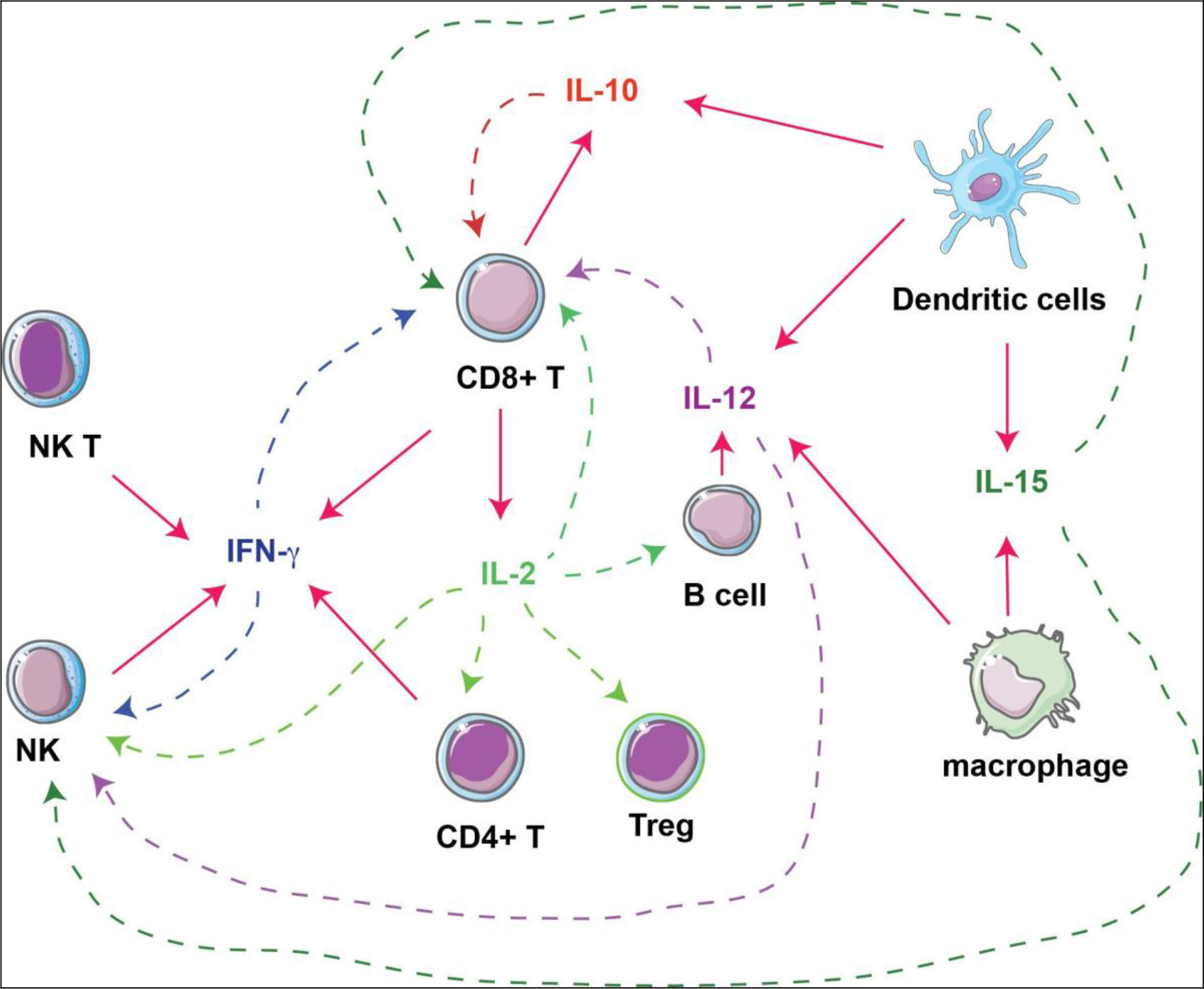
Figure 1. Interactions of cytokines and immune cells. Immunostimulatory cytokines produced by immune cells (solid arrows) are investigated as immunotherapies against tumors, by acting on different types of immune cells (dashed arrows).
References
- Gaffen SL, KD Liu, et al. (2004) Overview of interleukin-2 function, production and clinical applications. Cytokine 28: 109–23.
- Lotze MT, et al. (1985) In vivo administration of purified human interleukin 2. I. Half-life and immunologic effects of the Jurkat cell line-derived interleukin 2. J Immunol 134: 157–166.
- Rosenberg SA, et al. (1985) Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med 313: 1485–1492.
- Rosenberg SA, et al. (2014) IL-2: the first effective immunotherapy for human cancer. J Immunol 192: 5451–5458. [crossref]
- Burchill MA, Yang J, Vang KB, Farrar MA (2007) Interleukin-2 receptor signaling in regulatory T cell development and homeostasis. Immunol Lett 114: 1–8. [crossref]
- Charych, D, et al. (2017) Modeling the receptor pharmacology, pharmacokinetics, and pharmacodynamics of NKTR-214, a kinetically-controlled interleukin-2 (IL2) receptor agonist for cancer immunotherapy. PloS one 12: 0179431–0179431.
- Charych DH, et al. (2016) NKTR-214, an Engineered Cytokine with Biased IL2 Receptor Binding, Increased Tumor Exposure, and Marked Efficacy in Mouse Tumor Models. Clin Cancer Res 22: 680–690.
- Li Y, Strick-Marchand H, Lim AI, et al. (2017) Regulatory T cells control toxicity in a humanized model of IL-2 therapy. Nat Commun 8: 1762. [crossref]
- Lin F, Luo X, Tsun A, Li Z, Li D, et al. (2015) Kaempferol enhances the suppressive function of Treg cells by inhibiting FOXP3 phosphorylation. Int Immunopharmacol 28: 859–865. [crossref]
- Li Z, et al. (2014) PIM1 kinase phosphorylates the human transcription factor FOXP3 at serine 422 to negatively regulate its activity under inflammation. J Biol Chem 289: 26872–26881.
- Diab A, et al. (2018) NKTR-214 (CD122-biased agonist) plus nivolumab in patients with advanced solid tumors: Preliminary phase 1/2 results of PIVOT. J Clin Oncol 36(suppl; abstr 3006).
- Silva DA, Yu S, Ulge UY, et al. (2019) De novo design of potent and selective mimics of IL-2 and IL-15. Nature 565: 186–191. [crossref]
- Waldhauer I, et al. (2013) Novel Tumor-Targeted, Engineered IL-2 Variant (IL2v)-Based Immunocytokines For Immunotherapy Of Cancer. Blood 122: 2278.
- Lamichhane P, et al. (2017) IL10 Release upon PD-1 Blockade Sustains Immunosuppression in Ovarian Cancer. Cancer Res 77: 6667–6678.
- Emmerich J, et al. (2012) IL-10 directly activates and expands tumor-resident CD8(+) T cells without de novo infiltration from secondary lymphoid organs. Cancer Res 72: 3570–3581.
- Mumm JB, et al. (2011) IL-10 elicits IFNgamma-dependent tumor immune surveillance. Cancer Cell 20: 781–796.
- Naing A, Papadopoulos KP, Autio KA, Ott PA, Patel MR, et al. (2016) Safety, Antitumor Activity, and Immune Activation of Pegylated Recombinant Human Interleukin-10 (AM0010) in Patients With Advanced Solid Tumors. J Clin Oncol 34: 3562–3569. [crossref]
- Naing A, et al. (2018) PEGylated IL-10 (Pegilodecakin) Induces Systemic Immune Activation, CD8(+) T Cell Invigoration and Polyclonal T Cell Expansion in Cancer Patients. Cancer Cell 34: 775–791.
- Vignali DA, Kuchroo VK (2012) IL-12 family cytokines: immunological playmakers. Nat Immunol 13: 722–728. [crossref]
- Brunda MJ, Luistro L, Warrier RR, Wright RB, Hubbard BR, et al. (1993) Antitumor and antimetastatic activity of interleukin 12 against murine tumors. J Exp Med 178: 1223–1230. [crossref]
- Brunda MJ, et al. (1995) Role of interferon-gamma in mediating the antitumor efficacy of interleukin-12. J Immunother Emphasis Tumor Immunol 17: 71–77.
- Garris CS, et al. (2018) Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-gamma and IL-12. Immunity 49: 1148–1161.
- Rook AH, et al. (1999) Interleukin-12 therapy of cutaneous T-cell lymphoma induces lesion regression and cytotoxic T-cell responses. Blood 94: 902–908.
- Younes A, Pro B, Robertson MJ, Flinn IW, Romaguera JE, et al. (2004) Phase II clinical trial of interleukin-12 in patients with relapsed and refractory non-Hodgkin’s lymphoma and Hodgkin’s disease. Clin Cancer Res 10: 5432–5438. [crossref]
- Ansell SM, Witzig TE, Kurtin PJ, Sloan JA, Jelinek DF, et al. (2002) Phase 1 study of interleukin-12 in combination with rituximab in patients with B-cell non-Hodgkin lymphoma. Blood 99: 67–74. [crossref]
- Little RF, et al. (2006) Activity of subcutaneous interleukin-12 in AIDS-related Kaposi sarcoma. Blood 107: 4650–4657.
- Little RF, et al. (2007) Phase 2 study of pegylated liposomal doxorubicin in combination with interleukin-12 for AIDS-related Kaposi sarcoma. Blood 110: 4165–4171.
- Rudman SM, et al. (2011) A phase 1 study of AS1409, a novel antibody-cytokine fusion protein, in patients with malignant melanoma or renal cell carcinoma. Clin Cancer Res 17: 1998–2005.
- Fallon J, Tighe R, Kradjian G, Guzman W, Bernhardt A, et al. (2014) The immunocytokine NHS-IL12 as a potential cancer therapeutic. Oncotarget 5: 1869–1884. [crossref]
- Paoloni M (2015) Defining the Pharmacodynamic Profile and Therapeutic Index of NHS-IL12 Immunocytokine in Dogs with Malignant Melanoma. PLoS One 10: 0129954.
- Eckert F (2016) Enhanced binding of necrosis-targeting immunocytokine NHS-IL12 after local tumour irradiation in murine xenograft models. Cancer Immunol Immunother 65: 1003–1013.
- Xu C, et al. (2017) Combination Therapy with NHS-muIL12 and Avelumab (anti-PD-L1) Enhances Antitumor Efficacy in Preclinical Cancer Models. Clin Cancer Res 23: 5869–5880.
- Koneru M, et al. (2015) A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J Transl Med 13: 102.
- Conlon KC, et al. (2015) Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol 33: 74–82.
- Miller JS, et al. (2018) A First-in-Human Phase I Study of Subcutaneous Outpatient Recombinant Human IL15 (rhIL15) in Adults with Advanced Solid Tumors. Clin Cancer Res 24: 1525–1535.
- Mortier E, et al. (2006) Soluble interleukin-15 receptor alpha (IL-15R alpha)-sushi as a selective and potent agonist of IL-15 action through IL-15R beta/gamma. Hyperagonist IL-15 x IL-15R alpha fusion proteins. J Biol Chem 281: 1612–1619.
- Rhode PR, et al. (2016) Comparison of the Superagonist Complex, ALT-803, to IL15 as Cancer Immunotherapeutics in Animal Models. Cancer Immunol Res 4: 49–60.
- Romee R, et al. (2018) First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood 131: 2515–2527.
- Tang L, Zheng Y, et al. (2018) Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat Biotechnol 36: 707–716. [crossref]
- Harshman, LC, S Srinivas (2010) The bevacizumab experience in advanced renal cell carcinoma. OncoTargets and therapy 3: 179–189.
- Motzer RJ, et al. (2009) Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol 27: 3584–3590.
- Naito S, et al. (2010) Prognosis of Japanese metastatic renal cell carcinoma patients in the cytokine era: a cooperative group report of 1463 patients. Eur Urol 57: 317–325.
- Kaplan DH, et al. (1998) Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proceedings of the National Academy of Sciences of the United States of America 95: 7556–7561.
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, et al. (2017) Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 168: 542. [crossref]
- Karachaliou N, et al. (2018) Interferon gamma, an important marker of response to immune checkpoint blockade in non-small cell lung cancer and melanoma patients. Ther Adv Med Oncol 10: 1758834017749748.
- Buddingh EP, et al. (2015) Interferon-gamma Immunotherapy in a Patient With Refractory Disseminated Candidiasis. Pediatr Infect Dis J 34: 1391–1394.
- Nagai Y, et al. (2015) Disabling of the erbB Pathway Followed by IFN-gamma Modifies Phenotype and Enhances Genotoxic Eradication of Breast Tumors. Cell Rep 12: 2049–2059.
- Zhang H, et al. (2017) A targeted immunotherapy approach for HER2/neu transformed tumors by coupling an engineered effector domain with interferon-?. OncoImmunology, (just-accepted): p. 00–00.