DOI: 10.31038/MGJ.2022514
Abstract
Proteins present in most biological mixtures are expressed over a vast concentration range (up to 10-12 orders of magnitude in human sera), the most abundant ones complicating the detection of low-abundance species or trace components, Classical approaches, such as pre-fractionation and immuno-depletion methodologies are frequently used to remove the most abundant species. Unfortunately, these methods not only are unsuccessful in concentrating trace components, which could remain below the detection limits of analytical approaches, but also may cause a non-specifically depletion of other components (including low abundance ones). In case of immuno-depletion, the situation is less than brilliant: untargeted proteomic analyses using current LC-MS/MS platforms with immuno-depletion cannot be expected to efficiently discover low-abundance, disease-specific biomarkers in plasma, since the increment in detection of these trace components after such a treatment results in a meagre 25% increase, accounting for only 5-6%of total protein identifications in depleted plasma. The characterization of minor components in complex protein systems, has been revolutionized by the introduction of the combinatorial peptide ligand libraries technology. This new methodology is based on the use of hexa-peptide baits to capture and normalize the relative concentrations of the components of any proteome under investigation. The major advantage of this technique, in comparison with other pre-fractionation methods, is that it not only diminishes the concentration of the more abundant proteins, but also concentrates low-abundance and even trace components, thus providing access to the “invisible” proteome. In addition, the loss of low-abundance species that may be accidentally eliminated by co-depletion using immuno-subtraction methods, is avoided.
The Problems with Current Depletion Methods
The elimination of high-abundance proteins is operated by immunodepletion using specific solid-phase antibodies against the proteins to be suppressed. The method is quite effective; however, it suffers from a vicious circle which starts from the use of small volumes of expensive immunosorbents which accept only small samples to deplete. In small samples the amount of targeted markers is very low and they are additionally diluted during the process, thus rendering their detection even more challenging without any post concentration. Naturally concentration is possible but it contributes to protein losses. Immunosorbents are also limited for sample treatment due to their species specificity; immunosorbents available for proteomics are limited for the treatment of only human blood plasma. Conversely the use of enrichment methods based on solid-phase adsorption of targeted species or groups (e.g. glycoproteins, phosphoproteins and other classes) or the use of solid-phase combinatorial affinity ligands are by far more effective since they allow much larger initial biological samples and hence larger quantities of targeted low-abundance proteins. The Combinatorial Peptide Ligand Library (CPLL) is a technology for sample treatment that repeatedly demonstrated its capability to allow detecting proteins that are most of the time ignored because well below the level of sensitivity of proteomics equipments and methods. It is additionally of general use for various biological material and various species. This original procedure, that takes its origin from affinity chromatography mechanisms, when used under overloading conditions, contributes not only to improve the knowledge in proteomics but, more importantly, to detect dilute proteins that are expressed at the early-stage of metabolic diseases. It is after years of applications in various conditions and various sample situations that low-abundance protein detection by CPLL in early-stages of diseases is gaining momentum as a potential discovery allowing the design of diagnostic tools. Explanation of the mechanism of action is given in the following sections as well as examples of detection of panels of exclusive low abundance proteins present in various diseases [1,2].
Biomarker Discovery, a Major Target in Proteomics Investigations
This original procedure, that takes its origin from affinity chromatography mechanisms, when used under overloading conditions, contributes not only to improve the knowledge in proteomics but, more importantly, to detect dilute protein that are expressed at the early-stage of metabolic diseases. It is after years of applications in various conditions and various sample situations that low-abundance protein detection by CPLL in early-stages of diseases is gaining momentum as a potential discovery allowing the design of diagnostic tools. Explanation of the mechanism of action is given in the following sections as well as examples of detection of panels of exclusive low abundance proteins present in various diseases. Proteins and their variants are produced in a very large number and their individual concentration is extremely large, ranging throughout at least a dozen of orders of magnitude if not more. This situation renders the detectability of low- and very low-abundance species very challenging or clearly impossible in practice. Without any kind of sample treatment the large majority of proteins cannot be detected because their concentration is either below the detectability levels or because their signal is suppressed by the presence of most abundant proteins. the use of enrichment methods based on solid-phase adsorption of targeted species or groups (e.g. glycoproteins, phosphoproteins and other classes) or the use of solid-phase combinatorial affinity ligands are by far more effective since they allow much larger initial biological samples and hence larger quantities of targeted low-abundance proteins. The Combinatorial Peptide Ligand Library (CPLL) is a technology for sample treatment that repeatedly demonstrated its capability to allow detecting proteins that are most of the time ignored because well below the level of sensitivity of proteomics equipment and methods. This original procedure, that takes its origin from affinity chromatography mechanisms, when used under overloading conditions, contributes not only to improve the knowledge in proteomics but, more importantly, to detect dilute protein that are expressed at the early-stage of metabolic diseases. It is after years of applications that low-abundance protein detection by CPLL in early-stages of diseases is gaining momentum as a potential discovery allowing the design of diagnostic tools. Explanations of the mechanism of action is given in the following sections as well as examples of detection of panels of exclusive low abundance proteins present in various diseases.
The CPLL Capabilities to Detect Low-abundance Proteins from Early Stage Gene Expression
A combinatorial peptide ligand library (CPLL) is a quite recent technology now extensively described for successful applications in animal and plant proteomics investigations. Many applications have been reported with a major interest in the discovery of low- and very low-abundance proteins that are undetectable even after the use of immuno-depletion of major species. In practice the CPLL procedure allows compressing the dynamic concentration range of protein components by simultaneously decreasing the concentration of high-abundance species and enriching for low- and very low-abundance ones (for reviews see references This concept has been coined several years ago and since then had not reduced its interest for many applications including the discovery of markers of diagnostic and prognostic interest. The library is composed of millions of spherical gel porous beads each of them covalently carrying many copies of a single hexapeptide structure. The library is made via a combinatorial synthesis process that uses natural amino acids grafted the one after the other (split-and-pool procedure). Each bead can be considered as an affinity chromatography sorbent addressing a single or a group of proteins from the crude biological sample with a common affinity for the same peptide structure. Considering that the mixture of beads carries millions of different affinity beads, most, if not all, proteins are adsorbed. Under large overloading sample conditions, concentrated proteins (high abundance species) saturate rapidly the corresponding affinity beads while the excess remains free in solution. Conversely very dilute proteins (very low-abundance species) converge towards their specific beads and are thus concentrated. Upon completion of the binding process, dominated by not only adsorption, but also by quite intensive displacement effects, the beads are washed and all proteins in solution, mainly the excess of large abundance proteins, are eliminated. The adsorbed proteins are then desorbed using dissociation compounds such as those adopted in affinity chromatography; the collected sample thus comprises all captured proteins where their respective dynamic concentration range is much reduced. In this sample low-abundance proteins are detectable because first they are concentrated by the affinity process and also because their signal is not any longer obscured by the high-abundance species that are now largely diluted. The intense competition effect among proteins during the adsorption phase is the result of numerous molecular interactions singularly or collectively present generated by the mixed-mode affinity ligand library (the peptide). Among them are hydrophobic associations, electrostatic interactions and hydrogen bonding. The interaction forces are governed by the mass action law for systems that associate together by molecular affinity; the association and dissociation of partners depend on environmental conditions such as the pH, the ionic strength of the buffer, the temperature, the presence of competitors, their concentration and the extent of overloading. All these physicochemical parameters need to be considered with care in order to get the maximum reproducibility between samples. The two major success factors are (i) the enrichment of low abundance species, which is dependent on the availability of biological sample (the larger the sample, the higher level of enrichment) and (ii) the ability to desorb all proteins captured by the beads. In comparison to the so-called “depletion” or “immune-depletion” technologies, CPLLs show large distinctive characteristics. While depletion does not concentrate the low-abundance proteins, the CPLL main property is to concentrate most of very dilute species to bring them to the level of detectability by current analytical methods. High abundance proteins are not eliminated as this is the case with depletion methods, but rather maintained at a certain level of concentration conserving thus the property to carry other interacting polypeptides that are not The risk of protein losses due to non-specific binding on solid supports is prevented with CPLLs; adsorbed proteins necessitate a complete elution using various appropriate dissociation methods. Alternatively, after extensive washings the beads loaded with proteins can be directly trypsinized in order to produce peptides that are collected and streamlined within a LC-MS/MS equipment for protein identification.
Expected Upcoming Developments with CPLL
Recent technological developments about identifying early-stage modifications of protein expression for various critical pathologies are a promise for a great future. Statistical observations of low-abundance proteins expressed during the development of some cancers will improve the reliability of selection of marker candidates.
Post-translational modifications such as truncations, mis-glycosylations, mistaken phosphorylations and others, that are also tracked as potential biomarkers, could eventually be circumvented if the enzymes that are at the origin of such modifications are identified as very low-abundance proteins that are dependent on a bad or modified regulation of the expression system. Although the performance of CPLLs has largely contributed to the progress of novel discoveries, other complementary approaches associated to modern and more sensitive equipment, will increase the probability of novel reliable and affordable findings.
On CPLL technology itself possible developments are envisioned that could be advantageously associated to specific enrichment technologies. Thus the general enrichment governed by this multi-affinity principle could be enhanced by adding to the libraries various accurately selected adsorbents in order to either increase the low-abundance species or to further enrich a special group of proteins. Main principles for this approach were already suggested in 2015. The mode of use of CPLLs could also be progressively normalized as a function of the type of biological samples. For the discovery phase of novel biomarkers, two main general routes are used today: (i) the direct comparative expression difference of previously fluorescently labelled samples by 2DDIGE separation analysis and (ii) the indirect comparative tryptic digests of enriched samples, classified as bottom up approach.
Both are more rapid than other intricate protein capture methods and multiple sequential elutions from beads followed by technological clean-up and/or fractionation methods with the additional risk of protein losses. Although to date the return from massive efforts in proteomics is quite scarce in terms of diagnostic tests, the search for early-stage protein expression modifications continues. The acceleration of exploitable results in view of bringing findings to clinical practice is contingent upon deep collaborations between laboratories having complementary skills and also complementary interests including industrial organizations as well as bio-banks and clinicians. In this endeavour it is believed that CPLLs as are described or enriched by additional features may contribute to novel discoveries for early-stage potential protein biomarkers allowing differentiation of patients subgroups to fit with the current trends in personalized medicine.
Conclusions
In the early years of CPLL applications, we felt the growth of the methodology was in a stage of “Andante moderato”, like in the second theme of the third movement of the famous Symphony No. 9 by Ludwig van Beethoven (LvB). Yet, as the years went by, and as witnessed by the graph in Figure 1, it would appear that now CPLLs have reached the stage of “Andante maestoso”, as in the fourth movement of the Symphony, which ends with the Hymn to Joy from Friedrich Schiller (FS). It is hoped that more and more scientists will pick up the technique, given its high performance (Figure 2).
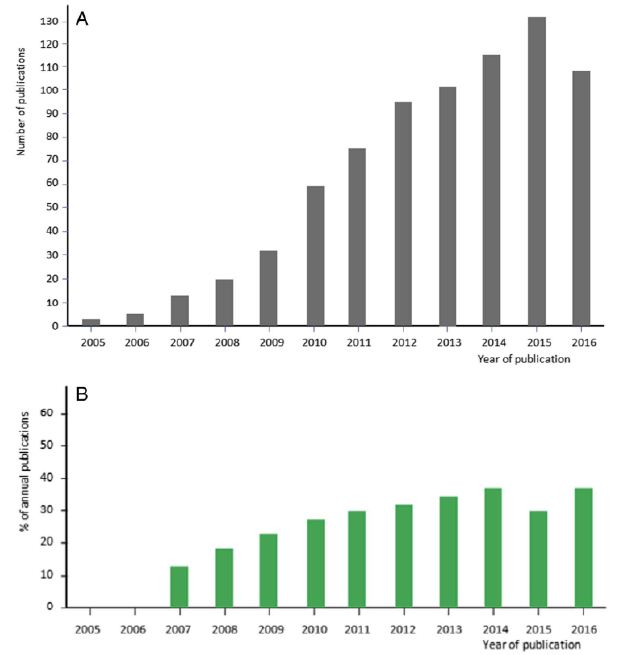
Figure 1: Progression of the number of publications over years mentioning the use of CPLL. A: representation of the number of published papers from 2005 to 2016. The last year represents an incomplete count. B: progression of published reports on CPLL evidencing or mentioning their use within the domain of biomarker discovery. Each bar is expressed in % of the total number of published papers shown above in panel A.
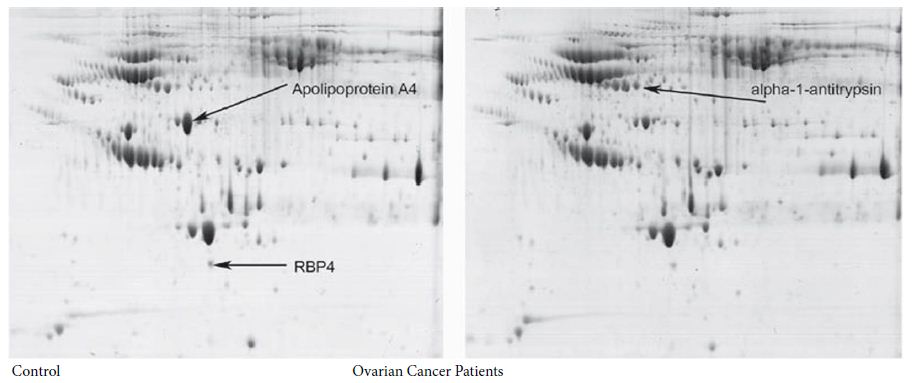
Figure 2: Two dimensional polyacrylamide gel electrophoresis of serum from healthy women (left panel) and from epithelial ovarian cancer (right panel). The first dimension separation was performed by using a relatively narrow pH gradient from 4 to 7. Both serum samples were treated by CPLL. Three spots of significantly different density between the groups were found (see arrowed indications) and then identified by MALDI mass spectrometry.
References
- Righetti PG, Boschetti E (2013) Combinatorial peptide libraries to overcome the classical affinity-enrichment methods in proteomics. Amino Acids 45: 219-229. [crossref]
- Boschetti E, Righetti PG (2013) Low-abundance Protein Discovery: State of the Art and Protocols, Elsevier. 978-0-12-401734-4, 2013.