Background
Hereditary haemoglobin disorders, also termed haemoglobinopathies, include mainly beta -thalassemia and sickle cell disease and represent the most common monogenic disorders in human. Cardiac complications are still a leading cause of mortality and morbidity in patients with haemoglobinopathy, have dramatically reduced in patient populations receiving modern regular therapy and follow-up.
Abstract
Τhe two main determinants of cardiovascular phenotype in haemoglobinopathy patients are the underlying molecular defect responsible for the main disease and the therapy applied for its management. The spectrum of cardiovascular manifestations in haemoglobinopathies is wide and includes ventricular dysfunction, pulmonary hypertension, thromboembolic events.
We report a case of thalassemia and one of sickle cell disease having different clinic phenotype with brief literature review.
Keywords
Heart failure, Hemoglobinopathies, Mortality
Case One
A patient 32-year-old male who was receiving periodic transfusions with intermittent chelation therapy for Cooley’s Anaemia came to our clinic with complaint of dyspnoea on exertion (DOE) in the past two years. The patient was followed up at the haematology department. He had a splenectomy at three years old. On the physical examination the patient had a global heart failure chart, high abundance ascites and hepatomegaly. He was in rapid atrial fibrillation. After treatment of congestion and slowing atrial fibrillation, the patient was explored by an echocardiography. Severe LV systolic dysfunction with a LVEF of about 20%; mitral regurgitation up to moderate degree was observed (Figure 1). The right ventricle is very dilated with massive and laminar tricuspid insufficiency due to lack of coaptation of the cusps thus creating a tricuspid hiatus. A restrictive filling pattern in both ventricles with both ventricular systolic dysfunctions were evident in this patient, also a lower right ventricular function: free wall TDI peak systolic velocity was 7 cm/sec. The patient was probed by MRI revealing advanced hepatic and cardiac hemochromatosis. Endocrine pancreas is also reached in view of high glycaemic dosage relating to secondary diabetes. Our patient died while in hospital because he had an electrical storm.
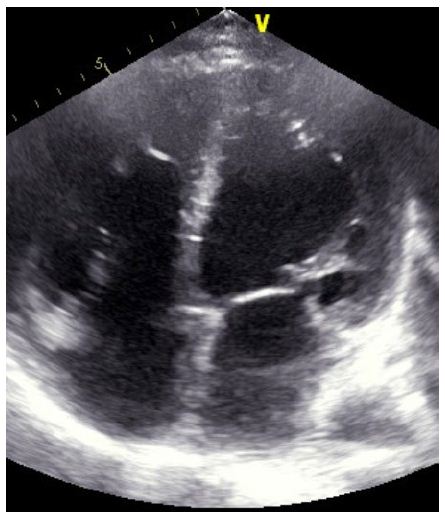
Figure 1: Echocardiography: four chamber 2D apical view : Biventricular dilated cardiomyopathy
Case Two
A patient 28-year-old male who was receiving periodic transfusions with intermittent chelation therapy for Sickle cell disease (SCD) came to our clinic with complaint of dyspnoea on exertion (DOE) in the past two years. The patient was followed up at the haematology department; she was on foldin, hydrea and vitamin C. On the physical examination the patient had only an enlarged jugular vein, high abundance ascites and hepatomegaly. At the electrocardiogram revealed a regular rhythm and a complete right bundle brunch block. At echocardiography, LVEF was about 60%. The right ventricle is much dilated (Figure 2) with massive tricuspid insufficiency due to a diastasis in tricuspid valve. A lower right ventricular function: free wall TDI peak systolic velocity was 4 cm/sec. Estimation of systolic pulmonary artery pressure from tricuspid insufficiency was about 120 mmHg which was very high and supra-systemic. The patient was explored by cardiac CT-scan revealing advanced pulmonary hemochromatosis (Figure 3), no pulmonary emboli and very enlarged right cardiac cavities. Our patient died of acute chest syndrome during hospitalization.
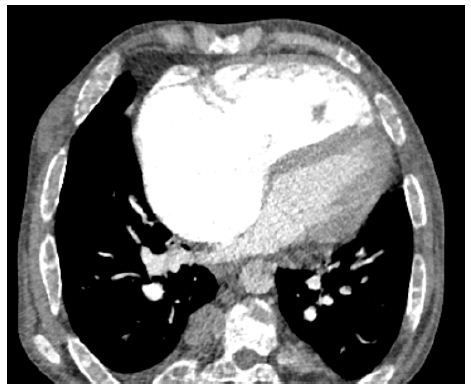
Figure 2: Cardiac CT scan : very dilated right ventricle in a SCD patient
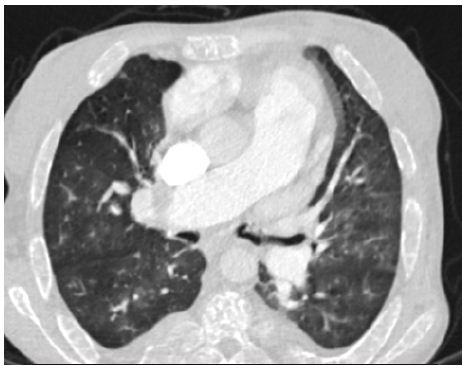
Figure 3: Pulmonary CT scan: Pulmoary hemochromatosis in a SCD patient
Discussion
Thalassemia heart disease involves mainly left ventricular dysfunction caused by transfusion-induced iron overload. In addition to the left ventricular abnormalities right ventricular dysfunction represents a common, yet less well explored complication in the cardiopulmonary spectrum of the disease. Biventricular dilated cardiomyopathy is still considered as the leading cause of mortality in patients with betathalassemia major [1,2]. In B-Thalassemia , the defect in haemoglobin is quantitative, characterized by a reduction or total depletion of 𝛽 chain synthesis, and the severity of 𝛽 chain deficiency determines the clinical phenotype, which extends from the severe and transfusion-dependent thalassaemia major to the milder and often transfusion-independent thalassaemia intermedia [3]. The heart takes up physiologic amounts of iron through transferrin receptors, but this process is tightly regulated and does not lead to iron overload. When transferrin-binding capacity is exceeded, circulating low molecular weight non-transferrin-bound iron (NTBI) species appear. NTBI is oxidatively active and can enter through nonspecific, poor-regulated cation channels in the heart, leading to cardiac iron overload [4]. Pulmonary hypertension (PH) in thalassemia is associated with vasoconstriction, vascular smooth muscle proliferation, and irregular endothelium in pulmonary arteries with associated thrombosis. These conditions all contribute to luminal narrowing, and eventual right ventricular failure. It includes plexiform and concentric medial hyperplastic pulmonary vascular lesions, and in situ pulmonary artery thrombosis [5,6]. These pulmonary vascular abnormalities may have resulted from chronic embolic disease in other patients [5]. Advancing age and a history of splenectomy are major risk factors for PH in this population [7,8]. Another phenomena can explains this phenomena as the process of haemolysis disables the arginine-nitric oxide pathway through the simultaneous release of erythrocyte arginase and cell-free haemoglobin Both nitric oxide and its obligate substrate arginine are rapidly consumed [9]. Outcome of heart failure, in advanced cardiac iron overload states, is dismal [10-12]. Compared to reported 3-month mortality rate of 58% in the pre-chelation era, recent findings indicate an improved prognosis over older series. Five-year survival was 48% and positively associated with left ventricular systolic function. All deaths occurred among patients with biventricular cardiomyopathy, shortly after involvement. Such improved survival is explained by the widespread use of chelation treatment and possibly also by better management of anaemia and use of angiotensin-converting enzyme inhibitors. Although LV Iron overload cardiomyopathy is a leading cause of death in patients with thalassaemia major this complication appears to be uncommon in SCD patients. Left ventricular dysfunction due to sickle cell disease is rare [13]. In SCD, the defect in haemoglobin is qualitative, as a substitution at the sixth amino acid residue in the 𝛽 chain results in synthesis of an abnormal haemoglobin, termed haemoglobin S, instead of the normal haemoglobin A [14]. Left ventricular systolic dysfunction is uncommon in patients with SCD: A meta-analysis of 19 controlled case studies has shown similar LV ejection fraction (LVEF) in homozygous S patients compared to healthy controls [15]. Accordingly, the prevalence of an LVEF < 50 % is low, ranging from 0% to 2.5% in ultrasound cohorts studies and 0% to 4% in smaller CMR studies [16,17]. SCD-related PH involves several mechanisms. First, as pulmonary pressure is the product of flow and pulmonary vascular resistances, high cardiac output in SCD induces elevated pulmonary pressure whether pulmonary vascular resistances are altered or not [18,19]. Second, chronic volume overload might lead to LV failure and subsequent pulmonary venous hypertension [20,21]. Third, intravascular haemolysis could induce pulmonary arterial vasculopathy mainly driven by nitric oxide scavenging due to free plasma haemoglobin [22]. Finally, several other mechanisms may participate including, chronic hypoxaemia, post-embolic PH, SCD-related lung injury, chronic liver disease, and asplenia.
In SCD, the pulmonary vascular bed is commonly affected [23]. Pneumonia may be difficult to distinguish from pulmonary infarction and both may coexist. Intravascular sickling may cause pulmonary vascular occlusion in the absence of radiologic changes, and in some patients bone marrow and fat released from infarcted bone may embolize to the lungs [24]. In the autopsy series of Gerry et al, 30% of adults and 22% of children had right ventricular hypertrophy. Three of these patients had had right ventricular failure, considered to be due to cor pulmonale [25].
Conclusion
The main cardiac involvement during major beta thalassemia is left ventricular dysfunction or in extreme cases biventricular dilated cardiomyopathy on the other hand sickle cell disease mainly causes right ventricular dilatation and dysfunction secondary to sickle cell lung pathology. A major near-term issue to address is the establishment of criteria for early disease-specific treatment of patients with MBT and SCD to avoid cardiovascular complications
Abbreviations
DOE: Dyspnoea on Exertion
MRI: Magnetic Resonance Imaging
SCD: Sickle Cell Disease
LVEF: Left Ventricle Ejection Fraction
MBT: Major Beta-thalassemia
References
- Grisaru D, Rachmilewitz EA, Mosseri M, Gotsman M, Lafair JS, et al. (1990) Cardiopulmonary assessment in beta-thalassemia major. Chest 98:1138‑42. [crossref]
- Levy RI, Moskowitz J (1982) Cardiovascular research: decades of progress, a decade of promise. Science 217: 121-12 [crossref]
- Rund D, Rachmilewitz E (2005) Beta-thalassemia. N Engl J Med 353: 1135-1146.
- Wood JC (2009) Cardiac complications in thalassemia major. Hemoglobin 33: 81-86.
- Sonakul D, Pacharee P, Thakerngpol K (1988) Pathologic findings in 76 autopsy cases of thalassemia. Birth Defects Orig Artic Ser 23: 157-176. [crossref]
- Morris CR, Gladwin MT, Kato GJ (2008) Nitric oxide and arginine dysregulation: a novel pathway to pulmonary hypertension in hemolytic disorders. Curr Mol Med 8: 620-632. [crossref]
- Atichartakarn V, Likittanasombat K, Chuncharunee S, Chandanamattha P, Worapongpaiboon S et al. (2003) Pulmonary arterial hypertension in previously splenectomized patients with beta-thalassemic disorders. Int J Hematol 78: 139-145. [crossref]
- Phrommintikul A, Sukonthasarn A, Kanjanavanit R, Nawarawong W (2006) Splenectomy: a strong risk factor for pulmonary hypertension in patients with thalassaemia. Heart Br Card Soc 92: 1467-1472. [crossref]
- Rother RP, Bell L, Hillmen P, Gladwin MT (2005) The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA 293: 1653-1662. [crossref]
- Engle MA, Erlandson M, Smith CH (1964) Late Cardiac Complications Of Chronic, Severe, Refractory Anemia With Hemochromatosis. Circulation 30: 698-705. [crossref]
- Engle MA (1969) Cardiac involvement in Cooley’s anemia. Ann N Y Acad Sci 119: 694-702. [crossref]
- Felker GM, Thompson RE, Hare JM, et al. (2000) Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N Engl J Med 342: 1077-1084. [crossref]
- Falk RH, Hood WB (1982) The heart in sickle cell anemia. Arch Intern Med 142: 1680-1684.
- Rees DC, Williams TN, Gladwin MT (2010) Sickle-cell disease. The Lancet 376: 2018-2031.
- Poludasu S, Ramkissoon K, Salciccioli L, Kamran H, Lazar JM (2013) Left ventricular systolic function in sickle cell anemia: a meta-analysis. J Card Fail 19: 333-341. [crossref]
- Desai AA, Patel AR, Ahmad H, Groth JV, Thiruvoipati T, et al. (2014) Mechanistic Insights and Characterization of Sickle Cell Disease Associated Cardiomyopathy. Circ Cardiovasc Imaging 7: 430-437. [crossref]
- Wood JC, Tyszka JM, Carson S, Nelson MD, Coates TD (2004) Myocardial iron loading in transfusion-dependent thalassemia and sickle cell disease. Blood 103: 1934-1936. [crossref]
- Mushemi-Blake S, Melikian N, Drasar E, Bhan A, Lunt A, et al. (2015) Pulmonary Haemodynamics in Sickle Cell Disease Are Driven Predominantly by a High-Output State Rather Than Elevated Pulmonary Vascular Resistance: A Prospective 3-Dimensional Echocardiography/Doppler Study. Connes P, éditeur. PLOS ONE 10: 0135472. [crossref]
- Caughey MC, Hinderliter AL, Jones SK, Shah SP, Ataga KI (2012) Hemodynamic Characteristics and Predictors of Pulmonary Hypertension in Patients with Sickle Cell Disease. Am J Cardiol 109: 1353-135 [crossref]
- Junqueira FP, Fernandes JL, Cunha GM, T A Kubo T, M A O Lima C, et al. (2013) Right and left ventricular function and myocardial scarring in adult patients with sickle cell disease: a comprehensive magnetic resonance assessment of hepatic and myocardial iron overload. J Cardiovasc Magn Reson 15: 83. [crossref]
- Fonseca GHH, Souza R, Salemi VMC, Jardim CVP, Gualandro SFM (2012) Pulmonary hypertension diagnosed by right heart catheterisation in sickle cell disease. Eur Respir J 39: 112-118. [crossref]
- Farmakis D, Aessopos A (2011) Pulmonary Hypertension Associated With Hemoglobinopathies: Prevalent But Overlooked. Circulation 123: 1227-1232. [crossref]
- Bromberg PA (1974) Pulmonary aspects of sickle cell disease. Arch Intern Med 133: 652-657
- Baroldi G (1969) High Resistance of the Human Myocardium to Shock and Red Blood Cell Aggregation (Sludge). Cardiology 54: 271-277. [crossref]
- Gerry JL, Bulkley BH, Hutchins GM (1978) Clinicopathologic analysis of cardiac dysfunction in 52 patients with sickle cell anemia. Am J Cardiol 42: 211-216. [crossref]