Abstract
Within classical nucleation theory (CNT), an initially formed crystal nucleus is often treated analogously to a droplet, with its evolution primarily described by changes in cluster size. However, increasing evidence suggests that the internal phase state of a cluster, including density for liquids and crystallinity for solids, evolves together with size and plays a decisive role in determining nucleation pathways and rates. Here we relate in situ transmission electron microscopy (TEM) observations of intermediate monomers to recent theoretical simulations, showing a phase-aware description of nucleation. Time-resolved TEM reveals that (i) coalescence proceeds through discrete, multistage events assisted by e-beam energy input; (ii) intermediate monomers repeatedly switch roles between reactants and products as their phase stability evolves; and (iii) mass redistribution can occur, consistent with crystallinity-dependent chemical potential and phase-dependent susceptibility to vaporization and redeposition. Motivated by these observations, we propose a more general framework that extends classical nucleation descriptions by incorporating coupled size and phase variables together with external driving fields. This phase aware description reconciles nonclassical multistage pathways with a tractable energetic model and suggests experimentally accessible control parameters, such as dose rate and dwell time, for steering early stage nanocrystal evolution.
Keywords
Nucleation, Nonclassical crystallization, In Situ TEM, Electron beam
Introduction
Understanding nucleation and early growth is central to controlling nanomaterial synthesis and transformation. Although classical nucleation theory (CNT) remains a useful baseline, its size-only description implicitly assumes that a nascent nucleus already possesses the bulk properties of the product phase. Recent rare-event simulations of Lennard–Jones vapor condensation demonstrate that this assumption can fail even for simple fluids: critical droplets nucleate with a density substantially different from the macroscopic liquid, and the reactive pathway involves simultaneous growth and densification [1]. In solids, an analogous internal variable is structural order (crystallinity, defect density, polymorph), which can evolve concurrently with size and invert size-only expectations such as anti-Ostwald ripening [2].
ZnO is a particularly suitable model to explore this coupling because it exhibits crystalline, poorly crystalline and amorphous states on the nanoscale, and these states respond differently to electron irradiation. In earlier in situ TEM studies, we observed multistage transitions among ZnO intermediate species and anti-Ostwald-like mass diffusion, indicating that intermediate monomers are not rigid building blocks but continually change size and phase [2]. More recently, we showed that e-beam irradiation can facilitate crystal reconstruction and even promote vapor-mediated mass transfer between unconnected particles, with the direction set by crystallinity- dependent stability [3].
In this paper, as an extension of our previous studies [2,3], we revisit representative multistage coalescence events in the pulsed- laser-produced ZnO system and use them to formulate a concise, phase-aware nucleation framework that bridges liquid condensation and nanocrystal evolution. Our goals are to distill experimental signatures of coupled size and phase evolution under e-beam driving, and to provide a minimal model that can be used to interpret and control such pathways in nanoscale crystallization.
Results and Discussion
Revisiting Classical Nucleation Theory
The theoretical fundamentals of crystal synthesis and crystallization are mainly based on classical theories, ascending to Gibbs free energy theories [4]. In CNT, when a crystal is initially formed, it could be regarded as a droplet. The Gibbs free energy ΔG of a droplet (assuming in a spherical shape) is described as in the following:
![]()
where ![]() is the bulk energy,
is the bulk energy, ![]() is the surface energy also referred to as the resistance force for nucleation, and r is the radius of the droplet.
is the surface energy also referred to as the resistance force for nucleation, and r is the radius of the droplet. ![]() is often referred to as the driving force of nucleation. The difference in bulk free energy between product and reactant is related to saturation of solution, which is expressed as:
is often referred to as the driving force of nucleation. The difference in bulk free energy between product and reactant is related to saturation of solution, which is expressed as:
where C is the concentration of a solution, C0 is the concentration of a solution when it is saturated, k is the Boltzmann constant, and T is temperature.
The Gibbs free energy change with the crystal radius is shown in Figure 1. A diagram of Gibbs free energy is provided vs. radius of nucleation, showing graphs of interface free energy, ![]() , the Gibbs free energy, ΔG, and bulk energy,
, the Gibbs free energy, ΔG, and bulk energy, ![]() . The maximum value of free energy is the energy barrier for nucleation, ΔG*, when
. The maximum value of free energy is the energy barrier for nucleation, ΔG*, when ![]() Correspondingly, the value of r* at the energy barrier is the critical size of nucleation, after which the addition of new molecules to nuclei decreases the free energy, so nucleation is favorable.
Correspondingly, the value of r* at the energy barrier is the critical size of nucleation, after which the addition of new molecules to nuclei decreases the free energy, so nucleation is favorable.
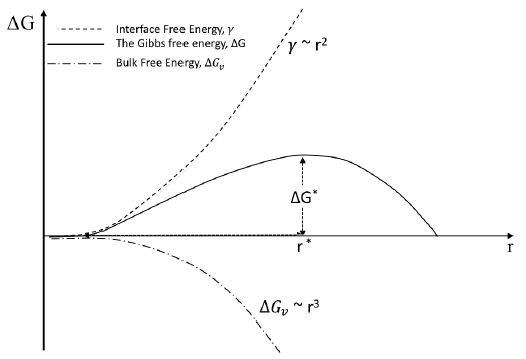
Figure 1: Free Gibbs energy diagram of nucleation in classical nucleation theory.
CNT remains a foundational framework because it reduces an intrinsically collective, many body process to a simple thermodynamic competition between a bulk free energy gain that scales with cluster volume and an interfacial free energy penalty that scales with cluster area. This reduction yields a single critical cluster size and a corresponding free energy barrier, providing a convenient and widely adopted basis for comparing nucleation behavior across different systems and conditions. However, the most consequential assumption underlying CNT is not the capillary approximation itself, but rather the implicit identification of the nucleating cluster with the equilibrium bulk phase. While cluster size quantifies the amount of material involved in nucleation, it does not specify the internal thermodynamic or structural state of that material.
In many nucleation processes, internal equilibration within the cluster is not instantaneous. Liquid droplets may initially form with reduced density and undergo subsequent densification, while crystalline nuclei may first appear as amorphous or poorly ordered aggregates before transforming into an ordered phase. Similarly, nanoparticles can nucleate in metastable polymorphs or defect rich configurations and later evolve toward more stable structures. When internal ordering and cluster growth occur on comparable time scales, nucleation cannot be adequately described by a single size parameter, but must instead be treated as a coupled process involving at least two collective variables: a size coordinate, such as the cluster radius R, particle number n, or volume, and an internal phase or order parameter, such as density ρ in fluids or crystallinity and structural order α in solids, with additional variables including composition or strain becoming relevant in more complex systems.
In literatures [5], the authors regarded the intermediate monomers, i.e., the primary particles, having energy at its minimum as ![]() , where
, where ![]() is its energy averaging over both surface and bulk energies. The intermediate monomers were proposed to contribute the free energy of the crystallization system by
is its energy averaging over both surface and bulk energies. The intermediate monomers were proposed to contribute the free energy of the crystallization system by ![]() . Therefore, the critical energy of the system change is
. Therefore, the critical energy of the system change is ![]() It implied that the primary particle was a precursor-like reactant, which has the effect equivalent to a change in the saturation of the solution, from S0Sp x S, where the supersaturation is defined as
It implied that the primary particle was a precursor-like reactant, which has the effect equivalent to a change in the saturation of the solution, from S0Sp x S, where the supersaturation is defined as ![]() , where C is the concentration and Cs is the solubility limit. As a result, the intermediate monomers affects the free energy of the system simply by adding/subtracting an amount of free energy (depending on the primary particles metastable/stable).
, where C is the concentration and Cs is the solubility limit. As a result, the intermediate monomers affects the free energy of the system simply by adding/subtracting an amount of free energy (depending on the primary particles metastable/stable).
In the model proposed by Mirabello and coworkers [6], the process of non-classical crystallization was described as a three-stage transition: (1) the formation particle (P); (2) the aggregation of Ps in which the Gibbs free energy changes due to the change of surface area; (3) the phase transition into the final crystalline phase. In the stage of the phase transition, a factor was assigned to describe the degree of the conversion. However, the definition of the conversion factor Φ is intuitional. Neither mass conservation nor additional nucleation was considered. Moreover, the model identified a critical cluster size, Rconv, by the condition: ![]() It implies that the transition direction is predetermined, from P to crystals (C) with increasing of Φ, and the process is size depended that in the early stage of R < Rconv the phase is dominated by P, and the conversion to C becomes favorable above Rconv. However, this predetermined phase change direction excludes the possibility of phase change from C to amorphous (A), or the possibilities of the growth of P consuming C, as we observed in this study.
It implies that the transition direction is predetermined, from P to crystals (C) with increasing of Φ, and the process is size depended that in the early stage of R < Rconv the phase is dominated by P, and the conversion to C becomes favorable above Rconv. However, this predetermined phase change direction excludes the possibility of phase change from C to amorphous (A), or the possibilities of the growth of P consuming C, as we observed in this study.
Observation of Multistages of Crystal Evolution
ZnO crystals or intermediate monomers in this study were produced by the pulsed laser method, similar to the previous studies [7]. The precursor solution for producing ZnO crystals was prepared from zinc acetate dihydrate in alcoholic solution under basic conditions [8]. Specifically, 1.5mM Zn(CH3COO)2 · 2H2O was dissolved in 25 mL DI water and mixed with 25mL ethanol (all chemicals from Sigma-Aldrich). Additionally, 1mL dilute NH3 · H2O solution (pH = 10) was added dropwise under vigorous stirring at room temperature. The substrate of Si (100) in a size of 1 cm × 1 cm was cleaned by DI water and immersed in the precursor solution. Copper grid holder for Transmission Electron Microscope (TEM) was placed on the silicon substrate, with film side face-up, and the grid edge was sealed by copper tape. The copper grid has 400 mesh with a thin film of pure silicon monoxide (15 – 30 nm) (SF400-Cu from Electron Microscopy Sciences, Hatfield, PA). The substrate was irradiated by pulsed laser for 1s. Ytterbium pulsed fiber laser with a wavelength of 1064 nm, pulse width of 100 ns was used to irradiate the substrate to trigger the hydrothermal reaction. Pulsed laser power density is 1.27 kW/cm2 in a repetition rate of 100kHz. After irradiated by the pulsed laser, the grid was taken out and rinsed with DI water. After dried by air, it was cleaned by plasma (Ar) for 40s before TEM observation. FEI Tecnai G2 20 TEM with 200 kV LaB6 filament was used. The CCD camera is a bottom mount Gatan US1000 2K x 2K, and videos were captured with a frame rate of ~30s-1 [frames per second (fps)].
Figure 2 shows an in-situ observation of multistages of crystallization. Starting from 1 s, two adjacent ZnO particles (intermediate monomers) can be resolved as distinct entities in the time resolved TEM sequence, each maintaining its own boundary and contrast. Under continuous electron beam irradiation, the pair evolves progressively toward integration through coupled interfacial and structural relaxation processes. As irradiation proceeds, surface atom diffusion smooths initially rough or high energy surface segments and promotes the development of a contact neck at the junction, while gradual lattice rearrangement and rotation reduce the crystallographic mismatch across the interface. Concurrently, local mass transport and boundary relaxation decrease the interfacial free energy, leading to a steady loss of boundary sharpness and a more coherent morphology. By the end of the 60 s observation window, the two monomers have merged into a single larger particle that exhibits a more uniform, crystal like contrast, consistent with the electron beam facilitated structural evolution mechanisms previously reported for multiphase nano ZnO, including surface diffusion, grain rotation, and irradiation activated mass transfer that collectively drive the system toward a lower energy configuration [3].

Figure 2: Particle coalescence processes. The frame size of each picture: w:153 nm, h:133 nm.
The theoratical model could be summarized in Figure 3 that in one stage of the reaction during the multi-stage process, from monomer (Mi) at stage i, which composed of two monomers (Mi1 and Mi2) to monomer (Mi+1) at stage i+1, representing the aggregated monomer (Mi1+Mi2). ΔE represents the energy barrier of particles’ moving, attaching, and aggregating under the assistance of the mass diffusion. The electron beam provided the energy for overcoming the energy barrier. Monomer Mi has a Gibbs energy of Gi, and monomer Mi+1 has a smaller energy of Gi+1. ΔG<0 indicates that Mi+1 is more stable than Mi. Monomer Mi+1 will continue interacting with surrounding monomers, evolving in the following stages.
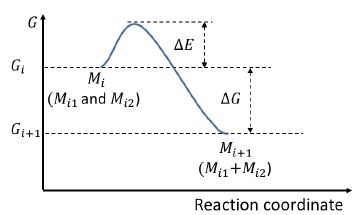
Figure 3: Energy diagram of the particle coalescence processes shown in Figure 2.
Mass Redistribution Among Clusters
Mass transfer among monomers was experimentally observed and shown as an important mechanism in the crystallization process. The continuous change of individual monomers (particles) indicated that successive mass transfer and phase change was induced by e-beam. Figure 4 was viewed from a TEM view window; each frame size is about 1.28× 0.81 μm. Two particles were tracked over 100s; the particle on the left is marked as p1, and the particle on the right is marked as p2. Two particles didn’t have significant change during the first 10s. At 12.90s, e-beam intensity increased artificially. As a result, particles shrank and p1 was hardly observed. At 22.83s, particles reappeared and continue to grow. At about 53.87s, p1 and p2 showed a crystalline structure rather than round shape. After 66.27s, particles continue to shrank. p1 disappeared at 73.93s and reappeared at 78.63s, and p2 disappeared at 78.63s. Although they disappeared eventually, a series of shrinkage and growth indicated the existence of local minimums in crystal growth energy diagrams.
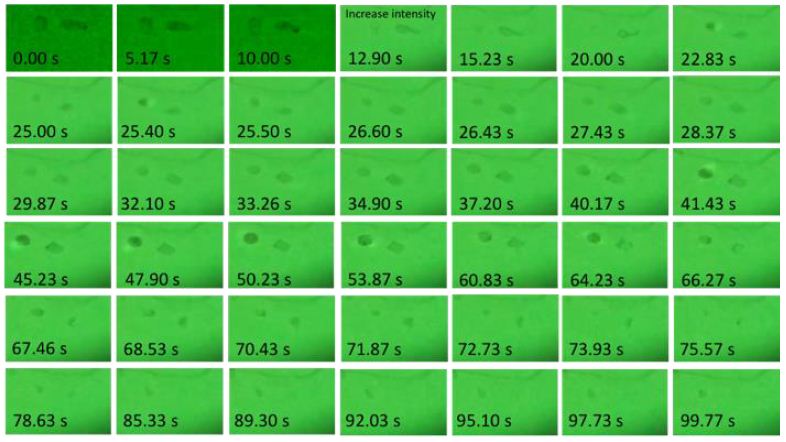
Figure 4: A series of phase changes of two particles due to e-beam. Snapshots from videos of observations from TEM view window. Magnification is 19.5k. Each frame size is about 1.28 × 0.81 μm. E-beam intensity increase at 12.90 s.
The reaction is found dose sensitive, in which a high dose rate leads to a high reaction rate. Particles didn’t have significant change until increasing the e-beam intensity (dose rate) at 12.90 s. In-situ measurement of the intensity of e-beam interacting with the sample is difficult (not allowed at our equipment). However, it could be a good reference using the dose rate projected at the screen, which records the electron density transmitted through the sample to detectors sitting below the sample. In this study, a dose rate of 673 e/(nm2s) was used at a magnification of 19.5 k; 1560 e/(nm2s) at a magnification of 29 k; and 3330 e/(nm2s) at a magnification of 43 k. Convert the electron flux to dose rate, ψ, on samples, using equation [9]:
![]()
where S is total stopping power for water, 2.79 (MeV cm2 g-1), a is the radius of irradiation area, I is the beam current, unit of Gy is defined as the absorption of one joule per kilogram of matter. Dose- related effect was studied by the effect of e-beam induced damage effect [10,11]. Therefore, the dose rate is then 3.01 × 107 Gy/s for 19.5 k, 6.98 × 107 Gy/s for 29 k, and 1.49 × 108 Gy/s for 43 k. Although it’s hard to have precise quantitative results between dose rate and crystallization kinetics, this result shows that electron beams could not only be used for imaging crystallization processes, but also as a promising tool to initiate and control the reactions in the observed area.
A General Framework of Crystallinity-dependent Nucleation Pathways
These observations align with our prior in situ TEM and simulation results on multiphase nano ZnO under electron irradiation, where the electron beam can induce constructive microstructural evolution rather than only damage [3]. Here, as an extension of our previous studies, we presented a generic model by extending the Gibbs energy profile of classical nucleation theories. The cases in ref. [5,6] discussed above could be included in this model. Moreover, two new features described by this model need to be stressed: firstly, considering that the crystal evolution results from the equilibrium among M, P, and C or A, intermediate monomers change their roles dynamically. Representative monomer types are schematically illustrated in Figure 5a. The relatively stable phase will consume less stable ones, e.g., the growth of stable P may consume adjacent C or A. Secondly, the phase transition and size change has no specific direction. The multi-stages reactions could result from either change of μMi , γMi or RMi , as long as the system reduce the Gibbs free energy as a whole, for example, a transition from A to C by reducing RMi .
The crystal evolution includes multi-stages reaction, and intermediate monomers at different stages indicated the local minimums in energy diagrams (shown in Figure 5b). Figure 5c describes a schematic overview of crystal evolution pathways, starting from intermediate monomers. Intermediate monomers were a mixture of ion-molecule monomers, dense liquid, amorous particles, poorly crystalline particles and nanocrystals, generated by the pulsed laser method from indirect nucleation in our system. The intermediate monomers under e-beam continue to undergo a kinetically favored pathway, in which mass- transfer induced new monomer formation and existed monomer dissolving lower energy barriers and alter evolution pathways.
In each stage (shown in Figure 5c), monomers move and attach, with or without adjustment of crystalline orientation depending on the phase properties of monomers, as discussed above. Then monomers coalesce directly or through mass distribution to form a new crystalline phase. The new crystalline phase then transforms into a more stable phase by mass redistribution and phase adjustment. Finally, the large newly formed phase shows as a more stable monomer, usually a crystalline phase, with lower Gibbs free energy and serves as the reactant for the next stage. The final crystal is the product of several stages of monomer interactions. In addition, as the trajectories of monomer interactions may not remain in the final crystals, single crystals could also be formed from various monomers’ interactions.
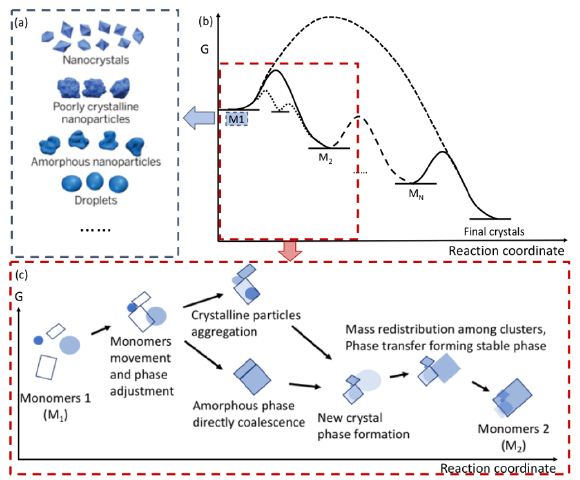
Figure 5: (a) Various monomers types shown in blue dash block, adapted from [12]. (b) Proposed crystallization dynamics schematic diagram showing multi-stages transformation from initial monomers M1 to final crystals. (c) A proposed example of a certain stage of crystal evolution. The mass and phase transfer from unstable phases to more stable phases. The product of a certain stage (M2) continue react and contribute to the crystal evolution in next stage.
The proposed mechanism in literatrues are certain possible crystal evolution pathways. Here, we extend the diversity of the intermediate reactions, in which the role of the primary particles or intermediate monomers could be more complex. The reaction from molecules (M) to crystals (C) or amorphous (A) involving primary particle (P) is not only in the form of M + P ![]() C or A as implied in ref. [5], but could be considered as the equilibrium among M, P and C or A as:
C or A as implied in ref. [5], but could be considered as the equilibrium among M, P and C or A as:
![]()
If P is unstable (Sp>1), reaction (1) will be reversely favored, and reaction (2)(3) is forwardly promoted. Similar to the case described in Figure S4D-E of Ref.5, consuming P will lower the total free energy of the system. However, if P is stable (Sp<1), we should consider whether it will still be consumed and increase the energy barriers, as described in Figure 3C-D of Ref. [5]. If it favors reaction in a different way, such as by the direct growth of P, the role of the primary particle will not always be a reactant but could be a product, and its effect on the free energy diagram of the system would be more complicated. The generic model proposed here implies all those possible roles of intermediate monomers and all the possible evolution pathways.
Conclusions
In summary, we combined in-situ intermediate monomer observations with a phase-aware nucleation viewpoint to highlight a unifying principle: nucleation and early growth are governed by coupled evolution of size and an internal phase coordinate. In the ZnO system, e-beam irradiation facilitates discrete multistage coalescence events, enables dynamic role switching of intermediates, and drives crystallinity-dependent, sometimes anti-Ostwald, mass redistribution. By casting these behaviors on a minimal two-coordinate free-energy landscape and including an explicit driving term for e-beam energy input, we obtain a compact framework that connects nanocrystal evolution to recently established variable-density nucleation pathways in liquid condensation. This framework motivates phase-aware experimental metrics and provides actionable control knobs for e-beam-assisted nanomanufacturing.
Notes: The authors declare no competing financial interest.
References
- Wu Y, Philippe T, Graini A, Lam J (2026) Nonclassical Nucleation Pathways in Liquid Condensation Revealed by Simulation and Phys Rev Lett. [crossref]
- Liu S, Liu C R (2021) Observation of New Dynamics of Transitions among Intermediate Species in Crystal Evolution and Its Role in a Generic Model of Journal of Physical Chemistry C. [crossref]
- Liu S, Wang R, Cai X, Wang Y (2025) Electron beam facilitated structural evolution of nano-zincoxide. Nanoscale. [crossref]
- Gibbs JW, Bumstead H A (1906) Longmans Green and Company
- Baumgartner J et al (2013) Nucleation and growth of magnetite from Nature Materials. [crossref]
- Mirabello G et al (2020) Crystallization by particle attachment is a colloidal assembly Nature Materials. [crossref]
- Liu S, Liu CR (2019) Morphology Control by Pulsed Laser in Chemical Deposition Illustrated in ZnO Crystal Crystal Growth and Design. [crossref]
- Pacholski C, Kornowski A, Weller H (2002) Self-Assembly of ZnO From Nanodots to Angew Chem Int Ed. [crossref]
- De Jonge N, Ross F M (2011) Electron microscopy of specimens in liquid. Nature Nanotechnology. [crossref]
- Egerton R F, Li P, Malac M (2004) Radiation damage in the TEM and Micron. [crossref]
- Jiang N (2015) Electron beam damage in oxides A Reports on Progress in Physics. [crossref]
- De Yoreo J J et al (2015) Crystallization by particle attachment in synthetic biogenic and geologic Science. [crossref]