DOI: 10.31038/CST.2022734
Synovial sarcoma is a malignant, soft tissue neoplasm of uncertain histogenesis demonstrating variable epithelial differentiation. Synovial sarcoma represents as a monophasic or biphasic malignant mesenchymal neoplasm characteristically demonstrating chromosomal translocation t(X;18) (p11; q11) incriminating genes SS18 and SSX1, SSX2 or SSX4. Neoplasm displays variable morphologies as monophasic spindle-shaped cell or monophasic epithelial cell or may manifest biphasic, myxoid, ossifying and poorly differentiated variants. Characteristic morphological features emerge as monotonous spindle-shaped cells permeated with vesicular, plump and overlapping nuclei. Cellular component is intermingled with haemangiopericytoma-like vascular articulations [1,2].
Median age of disease emergence is 35 years although no age of tumour emergence is exempt, and neoplasm may be discerned up to ninth decade (Figures 1 and 2).
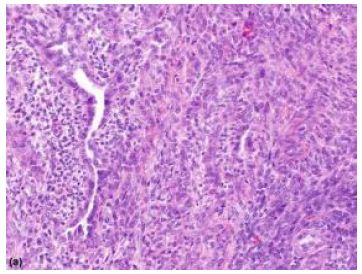
Figure 1: Synovial sarcoma depicting fascicles of spindle-shaped cells with uniform, vesicular, overlapping nuclei and minimal atypia. Branching, staghorn vascular articulations appear intermingled with neoplastic component (5).
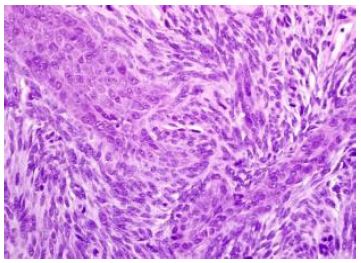
Figure 2: Synovial sarcoma composed of intersecting bundles of spindle-shaped cells within monotonous, uniform, overlapping nuclei and few mitotic figures (6).
Around ~18% instances occur within children and adolescents. A mild male predominance is observed with male to female proportion of ~1.2:1 [1,2]. Synovial sarcoma preponderantly incriminates extremities, trunk, head and neck, intrathoracic or intra-abdominal region or may arise within diverse locations although no site of tumour emergence is exempt. Soft tissue sarcomas depicting chromosomal translocation t (X;18; p11; q11) are contemplated to be synovial sarcomas. Synovial sarcoma is posited to occur due to chromosomal translocation t(X;18) (p11; q11) incriminating genes SS18 along with SSX1, SSX2 or SSX4 [1,2].
Majority (~90%) of synovial sarcomas manifest characteristic chromosomal translocation t(X;18) (p11.2; q11) along with SYT-SSX1 genetic fusion. Besides, chromosomal translocation t(X;18) (p11.21; q11) and SYT-SSX2 genetic fusion may occur [1,2]. Synovial sarcoma frequently depicts p16INK4A genetic deletion. Enhanced expression of EZH2 differentiates poorly differentiated neoplasms from monophasic or biphasic synovial sarcoma [1,2]. Aforesaid chromosomal translocation variably influences diverse oncogenetic pathways as initiation of SWI/SNF chromatin remodelling complex, polycomb repressor complex or canonical Wnt pathway. Partners of chromosomal translocation guide epithelial differentiation.
Chromosomal translocation SS18-SSX1 is preponderantly associated with monophasic synovial sarcoma (~70%) or biphasic neoplasms (~40%) whereas translocation SS18-SSX2 is predominant within monophasic tumours (~97%) and biphasic neoplasms (~3%). Translocation SS18-SSX1 prohibits Snail gene whereas amalgamation SS18-SSX2 prohibits Slug gene [1,2]. In contrast to Slug gene, intervention within Snail gene engenders intense de-repression of E-cadherin [1,2]. Intense expression of E-cadherin and extracellular matrix protein MMP2 functions as a prerequisite for occurrence of biphasic synovial sarcoma. Of obscure aetiology and pathogenesis, synovial sarcoma is posited to arise from multipotent mesenchymal stem cells or satellite cells as immature myoblasts [1,2].
Preceding radiation therapy exceptionally induces synovial sarcoma. Neoplastic concurrence with diverse syndromes or various diseases remains undocumented [1,2].
Grossly, synovial sarcoma represents as a soft to firm, well circumscribed or infiltrative, multinodular neoplasm. Upon initial representation, tumour magnitude varies from 3 centimetres to 10 centimetres although minute lesions < 1 centimetre diameter may occur, especially upon hands and feet. Cut surface is tan, grey/white, yellow, or pink. Foci of myxoid change necrosis, calcification and metaplastic ossification may be observed [1,2]. Upon microscopy, tumefaction exhibits mild cellular and nuclear pleomorphism [1,2].
Generally, neoplasm depicts subtypes as biphasic or monophasic, spindle-shaped or epithelial cell neoplasm [1,2]. Subcategories as monophasic epithelial, calcifying, ossifying, myxoid or poorly differentiated round cell synovial sarcoma may be exceptionally delineated [1,2]. Biphasic synovial sarcoma is constituted of dual components denominated by spindle-shaped cells and gland-like configurations of epithelial cells. Glandular lumens appear permeated with mucin. Neoplastic epithelial cells depict moderate, distinct amphophilic cytoplasm with spherical to elliptical nuclei. Squamous metaplasia can exceptionally occur [1,2]. Monophasic synovial sarcoma emerges as a hyper-cellular neoplasm with an infiltrative tumour perimeter. Fascicles of tumour cells appear intermingled with minimal stroma. Focal hyalinization or myxoid change is infrequent [1,2]. Neoplastic cells are monotonous and permeated with scanty, amphophilic cytoplasm, elliptical to spindle-shaped, vesicular nuclei with uniformly disseminated chromatin and inconspicuous nuclei. Indistinct nuclear palisading with nuclei overlapping adjacent nuclei may be exemplified [1,2].
Poorly differentiated synovial sarcoma is extensively cellular and is composed of spherical cells imbued with hyperchromatic nuclei. Mitotic activity is significant. Focal calcification and necrosis may ensue. Characteristically, focal staghorn vasculature, haemangiopericytoma-like foci or branching vascular configurations simulating solitary fibrous tumour may be observed. Mast cells are frequently discerned. Commonly discerned biphasic synovial sarcoma demonstrates commingling or distinct foci of dual neoplastic components. Metastatic neoplasms depict variable predominance of epithelial and spindle cell components [1,2].
Epithelial component commonly delineates enlarged; pale, columnar epithelial cells permeated with spherical, vesicular nuclei. Alternatively, cuboidal, flattened or spindle-shaped epithelial cells may occasionally be observed [1,2]. Epithelial cells may configure glandular articulations or tubules imbued with mucin. Papillary articulations or foci of squamous differentiation may exceptionally be discerned. Mucin rich variant of synovial sarcoma is infrequent [1,2]. Spindle cell component is composed of sheets or fascicles of miniature, uniform, plump, elongated cells pervaded with scanty cytoplasm, nuclei with dark, stippled nuclear chromatin and an indistinct cellular perimeter [1,2]. Tumour nodules or myxoid foci are infrequently discerned. Characteristically, intervening stroma enunciates thick, ropy collagen bundles. Commonly, stroma circumscribes cellular tumour nodules which appear intermingled with haemangiopericytoma-like vascular articulations. Foci of calcification may arise [1,2].
Monophasic synovial sarcoma is commonly composed of pure spindle-shaped cellular component demonstrating aforesaid morphological features [1,2].
Synovial sarcoma may minimally demonstrate a singular manifestation denominated as:
- Chromosomal translocation t (X;18; p11; q11)
- Immune reactivity to keratin
- Singular or multiple, aforesaid characteristic stromal features.
A pure epithelial cell component is negligibly observed (1,2). Frequently discerned nonspecific histological features occur as:
- Cellular palisading
- Pseudo rosettes
- Herringbone pattern
- Retiform or micro-cystic tumour configuration
- Occurrence of metaplastic bone or cartilage
- Foci of minimally enlarged cells.
Poorly differentiated synovial sarcoma may delineate a focal or pure, predominantly epithelial or mesenchymal tumour pattern [1,2].
Ultrastructural examination exhibits glandular configurations composed of epithelioid tumour cells demonstrating sparse luminal microvilli [1,2].
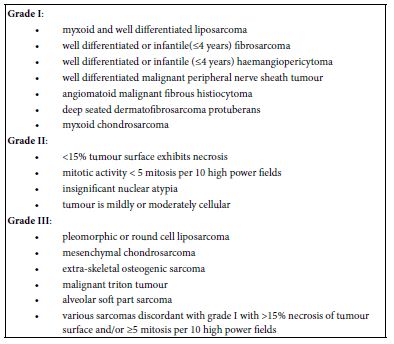
Staging of soft tissue sarcoma may be designated as:
- Stage I: Tumour is miniature and low grade as GX or G1
- Stage II: Tumour is miniature and high grade as G2 or G3
- Stage III: Tumour is enlarged and high grade G2 or G3
- Stage IV: Tumour dissemination into various body sites. Original tumour may be of variable magnitude (any T), any grade (any G) and may or may not demonstrate regional lymph node metastasis (any N) (2,3) (Table 1).
Table 1: Grading of non- rhabomyomatous paediatric soft tissue sarcoma as per Paediatric Oncology Group (POG) (2-3)
Synovial sarcoma is immune reactive to TLE1 and various cytokeratin wherein epithelial component is consistently immune reactive to CK7, CK8, CK14, CK18, CK19 and variably reactive to CK17, CK13, CK6 or CK16. Spindle cell component is focally and variably immune reactive to CK7, CK19, CK18, CK8, CK14, CK17 or CK20 [3,4]. Synovial sarcoma is immune reactive to epithelial membrane antigen (EMA), BCL2, β-catenin, calponin, CD99, CD56, CD57 or calretinin [3,4]. Epithelial component is comprehensively (100%) immune reactive to keratin or epithelial membrane antigen (EMA) whereas spindle cell component is immune reactive in ~80% instances [3,4]. Besides, neoplasm is immune reactive to SS18-SSX fusion specific antibody, SSX C terminus antibody or NY-ESO-1 [3,4]. Synovial sarcoma is immune non-reactive to CD34, desmin, h-caldesmon, myogenin, MyoD1, FLI1, WT1, SOX10, S100 protein or H3K27me3 [3,4].
Biphasic synovial sarcoma requires segregation from neoplasms such as adenocarcinoma, biphasic mesothelioma, glandular nerve sheath tumour, branchial analage mixed tumour or ectopic hamartomatous thymoma [3,4]. Monophasic synovial sarcoma necessitates demarcation from neoplasms as malignant peripheral nerve sheath tumour, cellular schwannoma, solitary fibrous tumour, leiomyosarcoma, spindle cell rhabdomyosarcoma, adult fibrosarcoma, dermatofibrosarcoma, protuberans with fibrosarcomatous transformation, epithelioid sarcoma, biphenotypic sinonasal sarcoma or sarcomatoid carcinoma [3,4]. Poorly differentiated synovial sarcoma mandates distinction from small round blue cell tumours as alveolar rhabdomyosarcoma, Ewing’s sarcoma, undifferentiated round cell sarcoma or Ewing-like sarcoma. Characteristic genomic modifications may be appropriately discerned upon reverse transcription polymerase chain reaction (RT-PCR). Cogent tissue sampling is mandated for confirmation of synovial sarcoma [3,4].
Corroborative tumour detection may be obtained with molecular or cytogenetic assessment for SS18-SSX genetic fusion with fluorescent in situ hybridization (FISH), reverse transcription polymerase chain reaction (RT-PCR) or next generation sequencing (NGS) [3,4]. Plain radiographs depict a site-specific spherical to elliptical, lobulated tumour mass. Incrimination of bone is infrequent [3,4]. Ossifying synovial sarcoma typically delineates spotty radio-opacities engendered due to focal calcification. Comprehensive surgical resection of neoplasm is a recommended therapeutic strategy [3,4]. Additionally, radiotherapy or adjuvant chemotherapy can be employed to manage challenging clinical scenarios as neoplasms >5 centimetre magnitude or tumours unamenable to surgical resection [3,4]. Adjuvant radiation therapy ameliorates prognostic outcomes and overall disease survival [3,4].
Neoplasms subjected to preceding radiation therapy may demonstrate moderate cellular and nuclear pleomorphism. Adjuvant chemotherapy can be beneficially adopted to treat neoplasms of advanced grade or tumours unamenable to pertinent therapy [3,4]. Agents such as ifosfamide or novel therapies as tyrosine kinase receptor inhibitor pazopanib or EZH2 inhibitor tazemetostat can be beneficially employed for therapeutic purposes. Also, T cell receptor-based immunotherapy implicating NY-ESO-1 within subjects demonstrating HLA-A*0201 is associated with clinical and radiological neoplastic melioration [3,4].
Inferior prognostic outcomes are associated with:
- SS18-SSX1 chromosomal translocation
- Monophasic and poorly differentiated synovial sarcoma
- Incriminated males
- Initial disease representation in elderly subjects
- Tumour magnitude ≥5 centimetres
- Neoplasm confined to centric, non-extremity sites
- Deep seated neoplasms
- Extensive tumour necrosis
- Mitotic activity ≥ 10 per high power field
- Elevated Ki67 proliferative index
- Precise tumour grade contingent to Fédération Nationale des Centres de Lutte Contre le Cancer (FNCLCC), as defined in incriminated adults
- Immunoreactivity to CXCR4 and IGF1R
- Tumour discernible within resected surgical perimeter
Expression of H3K27me3 and VEGF in concurrence with histological grade and stage of neoplasm along with emergence of distant metastasis (3,4).
References
- Manizhe AK, Mohseni I et al. 2022 Recurrent primary intracranial synovial sarcoma, a case report and review of the literature. Clin Case Rep 10. [crossref]
- Eliason L, Grant L et al. 2022 Qualitative study to characterize patient experience and relevance of patient-reported outcome measures for patients with metastatic synovial sarcoma. J Patient Rep Outcomes. 6. [crossref]
- Fice M, Almajnooni A et al. 2022 Does synovial sarcoma grade predict oncologic outcomes, and does a low-grade variant exist? J Surg Oncol. 125: 1301-1311. [crossref]
- Ren MY, Li J et al. 2022 Primary orbital monophasic synovial sarcoma with calcification: A case report. World J Clin Cases 10: 1623-1629. [crossref]