Abstract
Objective: A novel Cdk9 inhibitor 1-(2-adamantane-1-yl-1H indole-5-yl)-3-substituted thiourea derivative was designed and synthesized and their anti-gastric cancer activities were studied.
Methods: A series of target compounds 7a-7m were synthesized from amantadane formyl chloride by 6-step reactions. The structures of the target compounds were identified by 1H NMR, 13C NMR and HRMS. MTT assay was used to detect the inhibitory effect of synthetic compounds on the growth of gastric cancer cells, and Western blot assay was used to detect the regulatory effect of hit compound on downstream signaling pathways. Results: The results showed that the target compounds had certain inhibitory activity on the growth of gastric cancer cells, among which compound 7l had the best activity on the gastric cancer cell line (SGC-7901) with IC50 value of 2.26 ± 0.04 μM, and 7I had little toxicity on normal gastric epithelial cells (IC50 > 100 μM). In addition, 7I can bind to CDK9 and inhibit pSer2 expression in gastric cancer cells in a concentration dependent manner. Finally, molecular docking study showed that 7l can stably bind to the active site of CDK9 and has a high binding affinity.
Conclusion: This series of compounds has good anti-gastric cancer activity and has the significance of further study.
Keywords
Adamantane derivatives; Indole; Gastric cancer; CDK9
Gastric cancer is one of the most common cancers worldwide and ranks as the third leading cause of cancer-related deaths [1]. Middle-aged and elderly individuals are at high risk, and gastric cancer is often diagnosed at an advanced stage, with a five-year survival rate of less than 30% [2,3]. Therefore, understanding the occurrence and progression of gastric cancer, along with the potential discovery of novel diagnostic and prognostic biomarkers, is crucial for improving clinical outcomes. Clinically, the primary treatment for gastric cancer is surgical intervention, supplemented by pharmacological therapy [4]. Although chemotherapy can extend the survival of patients to some extent, conventional chemotherapeutic agents suffer from poor specificity, significant toxicity, side effects, and a tendency to induce drug resistance, all of which adversely affect treatment efficacy and patient survival [5]. Thus, identifying effective drugs with favorable therapeutic profiles for gastric cancer is of great importance. Cyclin-dependent kinases (CDKs) are a family of serine/threonine protein kinases that play vital roles in cell cycle progression and transcriptional regulation [6-9]. Recent studies have shown that CDKs are overexpressed in various cancers, leading to uncontrolled cell proliferation and drug resistance [10]. CDK9, a key member of the CDK family, is essential for stable RNA transcriptional elongation. CDK9 and cyclin T form the positive transcription elongation factor b (P-TEFb) complex, which promotes transcriptional elongation through phosphorylation of RNA polymerase II (RNAPII) [11,12]. Recent evidence indicates that CDK9 plays a critical role in numerous human cancers, including cervical, prostate, and lung cancers [13-18]. Dual-luciferase reporter assays have demonstrated that miR-613 inhibits the metastasis and progression of gastric cancer cells by downregulating CDK9 gene expression [19]. Therefore, targeting CDK9 to interfere with the development and progression of gastric cancer holds therapeutic promise. Building on previous work, this study designed and synthesized a novel series of CDK9 inhibitors (Figure 1). The influence of different substituted R groups on the antitumor activity of these thiourea compounds was investigated to identify more potent 1-(2-(adamantan-1-yl)-1H-indol-5-yl)-3-substituted thiourea derivatives. The synthetic route for target compounds 7a–7l is illustrated in Figure 1.

Figure 1: Synthetic route of compounds 7a~7l.
Materials and Methods
Chemicals and Reagents
Starting materials were purchased from Shanghai Titan Scientific Co., Ltd. All reagents were of analytical grade and used without further purification. Solvents were used as received. Thin-layer chromatography (TLC) silica gel (Qingdao Marine Chemical Factory, 60–100 mesh) and MTT powder (ST1537, Beyotime) were used.
Instruments
NMR spectra were recorded on a Bruker Avance 600 spectrometer using DMSO-d6 or CDCl3 as the solvent with TMS as the internal standard. Mass spectra were obtained using an Agilent 6230 mass spectrometer. A Thermo microplate reader was used for absorbance measurements.
Methods
MTT Assay for Cell Viability
The MTT assay was used to assess cell survival and growth. Cells were digested and seeded into 96-well plates at a density of 3,000 cells per well. After cell attachment, they were treated with the respective compounds or solvent, with cisplatin used as a positive control. After 48 hours of incubation, the medium was replaced with fresh medium containing MTT solution (1.2 mg/mL), and the cells were incubated for another 3 hours. The MTT formazan product was dissolved in DMSO, and the absorbance was measured at 492 nm using a microplate reader.
CDK9 Inhibition Assay
The ADP-Glo Kinase Assay was employed to determine the inhibitory activity of the compounds against CDK9 protein, conducted by Innovative CRO + Explorer (Beijing, China). Kinase activity was assessed in a buffer containing 50 mM Hepes, 10 mM MgCl2, 0.01% Brij 35, 1 mM EGTA, 2 mM DTT, and ddH2O. Test compounds were prepared in DMSO. The inhibition rate against CDK9/cyclin T1 (ATP: 20 μM) was tested for all compounds at a concentration of 1 μM.
Molecular Docking
Molecular docking was performed to explore the interaction mode between compound 7l and CDK9. The crystal structure of CDK9/cyclin T1 (PDB ID: 7NWK, resolution: 2.81 Å) was downloaded from the Protein Data Bank (https://www.rcsb.org/structure/7NWK). The protein structure was prepared using the Protein Preparation Wizard module in Schrödinger software (V 2023-1) under default parameters, including removal of water and solvent molecules, addition of hydrogen atoms, assignment of charges, completion of missing amino acid residues, optimization of the hydrogen bond network, and energy minimization using the OPLS4 force field. The structure of 7l was prepared using LigPrep (Schrödinger, LLC, New York, NY, 2023) under the OPLS4 force field with default parameters, including hydrogen addition, charge assignment, and generation of possible protonation states at pH 7.0 ± 2.0. Molecular docking was performed using the Ligand Docking module in Schrödinger software, docking 7l into the ATP-binding site of CDK9. The docking box was centered on the centroid of the original crystal ligand, and extra precision (XP) docking was employed. The binding energy of the 7l-CDK9 complex was calculated using the MM-GBSA module. Maestro (Version 12.5, Schrödinger) was used for visualization, and PyMOL (Version 2.5.4, Schrödinger, LLC) was used for rendering and presentation.
Experimental Methods and Results
Synthesis of Intermediate N-(o-Tolyl)adamantane-1-Carboxamide (2)
In a dried 50 mL round-bottom flask, toluene (10 mL), adamantane-1-carbonyl chloride (0.99 g, 5 mmol), o-toluidine (0.54 g, 5 mmol), and potassium carbonate (0.69 g, 5 mmol) were added successively. The mixture was stirred and heated to 80°C, and the reaction progress was monitored by TLC. After completion, the reaction mixture was cooled, filtered, washed with water, and dried to afford a crude white solid, which was recrystallized from ethanol to yield N-(o-tolyl)adamantane-1-carboxamide (1.17 g, 87%). 1H-NMR(600 MHz, CDCl3) δ: 7.88 (d, J=8.1 Hz, 1H), 7.24-7.18 (m, 2H), 7.17 (d, J=7.5Hz, 1H), 7.05 (dt, J=1.1, 7.4 Hz, 1H), 2.26 (s, 3H), 2.11 (brs, 3H), 1.99 (d, J=2.6 Hz, 6H), 1.77 (m, 6H).
Synthesis of Intermediate 2-(Adamantan-1-yl)-1H-indole (3)
A dried 25 mL three-necked flask was charged with N-(o-tolyl)adamantane-1 carboxamide (0.285 g, 1.0 mmol) and THF (15 mL). The mixture was cooled to 0–5°C under nitrogen, and n-butyllithium (2 mL) was added dropwise. The reaction was monitored by TLC. Upon completion, the pH was adjusted to 7.0–7.5 with 5% dilute hydrochloric acid. The mixture was extracted, and the organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography (eluent: petroleum ether/ethyl acetate=10: 1, v/v) to yield 2-(adamantan-1-yl)-1H-indole (0.166 g, 66.1%). 1H-NMR (600 MHz, CDCl3) δ: 7.30 (d, J=7.9 Hz, 1H), 7.18 (dd, J=0.5, 8.1 Hz, 1H), 6.88 (dt, J=1.1, 7.5 Hz, 1H), 6.83-6.77 (m, 1H), 5.98 (d, J=0.6 Hz, 1H), 1.97 (d, J=2.6 Hz, 3H), 1.93 (d, J=2.9 Hz, 6H), 1.73 (brs, 6H).
Synthesis of Intermediate 2-(Adamantan-1-yl)-5-nitro-1H-indole (4)
A 25 mL three-necked flask was charged with 2-(adamantan-1-yl)-1H-indole (0.25 g, 1.0 mmol) and concentrated sulfuric acid (0.8 mL). A solution of sodium nitrate (0.085 g, 1.0 mmol) in sulfuric acid (0.8 mL) was added dropwise at 0°C. After addition, the reaction was maintained for 1–2 hours and monitored by TLC. Upon completion, a large amount of ice water was added, and a yellow solid precipitated. The solid was filtered, washed with water, and dried. The crude product was purified by column chromatography (eluent: petroleum ether/ethyl acetate=5: 1, v/v) to yield 2 (adamantan-1-yl)-5-nitro-1H-indole (0.24 g, 82.8%) as a yellow solid. 1H-NMR (600 MHz, DMSO-d6) δ: 11.65 (brs, 1H), 8.44 (d, J=2.2 Hz, 1H), 7.93 (dd, J=2.2 ,9.0 Hz, 1H), 7.44 (d, J=8.8 Hz, 1H), 6.38 (d, J=1.5 Hz, 1H), 2.07 (brs,3H), 1.98 (brs, 6H), 1.81-1.67 (m, 6H);13C-NMR (150 MHz, DMSO-d6) δ: 154.0, 140.8, 139.9, 127.7, 116.9, 116.3, 111.4, 98.2, 41.9, 36.6, 34.1, 28.2; HRMS (ESI) m/z: 297.1599 [M+H]+, Calt: 297.1598 [M+H]+.
Synthesis of Intermediate 5-Amino-2-(adamantan-1-yl)-1H-indole (5)
In a 250 mL three-necked flask, ethanol (100 mL), acetic acid (10 mL), water (20 mL), and iron powder (8.62 g, 0.15 mol) were added. The mixture was heated to 75°C, and 2-(adamantan-1-yl)-5-nitro-1H-indole (11.4 g, 0.038 mol) was added in portions. The reaction was maintained for 2 hours and monitored by TLC. After completion, the reaction mixture was hot-filtered, and the filtrate was concentrated under reduced pressure to remove the solvent. Water (80 mL) was added, followed by ethyl acetate (80 mL) with stirring. The pH was adjusted to 7–8 with sodium bicarbonate. The organic phase was separated, dried over anhydrous Na22SO44, filtered, and purified by column chromatography (eluent: petroleum ether/ethyl acetate=2: 1, v/v) to yield 5 amino-2-(adamantan-1-yl)-1H-indole (6.9 g, 67.0%) as a red solid. 1H-NMR (600 MHz, CDCl3) δ: 7.80 (brs, 1H), 7.10 (d, J=8.4 Hz, 1H), 6.86 (d, J=2.2 Hz, 1H), 6.57 (dd, J=2.2, 8.4 Hz, 1H), 6.05 (dd, J=0.7, 2.2 Hz, 1H), 3.23-3.65 (m, 2H), 2.09 (brs, 3H), 1.96 (d, J=2.38 Hz, 6H), 1.78 (q, J=12.2 Hz, 6H);13C-NMR (150 MHz, CDCl3) δ: 149.9,139.3, 130.3, 129.4, 111.6, 110.8, 105.3, 95.5, 42.6, 36.8, 33.7, 28.5; HRMS (ESI) m/z: 267.1858 [M+H]+, Calt: 267.1856 [M+H]+.
Synthesis of Intermediate 5-Isothiocyanato-2-(adamantan-1-yl)-1H-indole (6)
In a dried 100 mL three-necked flask, a solution of 5-amino-2-(adamantan-1-yl)-1H indole (4.10 g, 15.5 mmol) in toluene (20 mL) and triethylamine (3 mL) was added. Carbon disulfide (3.50 g, 46.5 mmol) was added dropwise slowly. The reaction was stirred at room temperature for 8 hours. The mixture was filtered and dried, then dissolved in dichloromethane (30 mL). Triphosgene (5.0 g, 17 mmol) was added dropwise at 0–5°C. After 2 hours, the reaction was monitored by TLC. Upon completion, the product was purified by column chromatography (eluent: petroleum ether/ethyl acetate=10: 1, v/v) to yield 5-isothiocyanato-2-(adamantan-1-yl)-1H-indole (2.89 g, 64.0%) as a white solid. 1H-NMR(600 MHz, CDCl3) δ: 7.40 (d, J=1.8 Hz, 1H ), 7.23 (d, J=8.4 Hz, 1H, ), 6.98 (dd, J=2.0, 8.4 Hz, 1H), 6.20 (d, J=1.5 Hz, 1H ), 2.14 2.08 (m, 3H), 1.99-1.94 (m, 6H), 1.85-1.74 (m, 6H); 13 C-NMR (150 MHz, CDCl3) δ: 151.4, 134.1, 131.8, 128.7, 122.6, 119.0, 117.3, 111.2, 96.8, 42.5, 36.7, 33.9, 28.4; HRMS (ESI) m/z: 309.1422 [M+H]+, Calt: 309.1420 [M+H]+.
Synthesis of 1-(2-(Adamantan-1-yl)-1H-indol-5-yl)-3-Substituted Thiourea Derivatives (7a–7l)
In a 25 mL reaction flask, 5-isothiocyanato-2-(adamantan-1-yl)-1H-indole (0.145 g, 0.5 mmol) and toluene (10 mL) were added. The appropriate amine (0.5 mmol) was added, and the mixture was heated to 80°C. The reaction was monitored by TLC. After cooling, the mixture was filtered and dried to afford the target compounds as white solids in yields of 70.0–88.6%.
7a: white solid, yield 70.1%, mp 198~200℃, 1H NMR (400 MHz, DMSO-d6) δ: 10.92 (s, 1H), 9.25 (brs, 1H), 7.27 (dd, J=3.2, 4.95 Hz, 2H), 6.83 (dd, J=1.7 8.5 Hz, 1H), 6.08 (d, J=1.71 Hz, 1H), 3.47-3.40 (m, 2H), 2.06 (brs, 3H), 1.96 (d, J=1.96 Hz, 6H), 1.76 (br, 6H), 1.54-1.41 (m, 2H), 1.32-1.19 (m, 6H), 0.87 (t, J=6.79 Hz, 3H);13C NMR (100 MHz, DMSO-d6): δ 181.0, 152.5, 151.1, 135.3, 134.6, 128.4, 116.8, 111.5, 95.8, 44.5, 42.2, 36.8, 33.9, 31.5, 29.1, 28.3, 26.5, 22.5, 14.4; HRMS (ESI) m/z: 410.2625 [M+H]+, Calt: 410.2624 [M+H]+.
7b: white solid, yield 78.4%, mp 201~203℃, 1H NMR (400 MHz, DMSO-d6) δ: 10.90 (s, 1H), 9.21 (s, 1H), 7.32 (s, 1H), 7.25 (d, J=8.44 Hz, 1H), 6.96 (brs, 1H), 6.87 (dd, J=1.7, 8.4 Hz, 1H), 6.07 (d, J=1.3 Hz, 1H), 2.06 (brs, 3H), 1.96 (brs, 6H), 1.89-1.83 (m, 2H), 1.80-1.71 (m, 6H), 1.64 (d, J=12.6 Hz, 2H), 1.54 (d, J=12.7 Hz, 1H), 1.32-1.04 (m, 5H); 13C NMR (100 MHz, DMSO-d6) δ: 180.0, 151.0, 134.4, 130.1, 128.3, 118.8, 116.4, 111.3, 95.7, 52.9, 42.2, 36.8, 33.9, 32.4, 28.4, 25.6, 25.1; HRMS (ESI) m/z: 408.2469 [M+H]+, Calt: 408.2468 [M+H]+.
7c: white solid, yield 80.1%, mp 221~223℃, 1H NMR (400 MHz, DMSO-d6) δ: 10.91 (s, 1H), 9.66 (s, 1H), 9.35 (brs, 1H), 7.50 (d, J=7.8 Hz, 2H), 7.42 (s, 1H), 7.33-7.25 (m, 3H), 7.13-7.07 (m, 1H), 6.98 (dd, J=1.8, 8.5Hz, 1H), 6.10 (d, J=1.6Hz, 1H), 2.07 (brs, 3H), 1.97 (s, 6H), 1.84-1.66 (m, 6H); 13C NMR (100 MHz, DMSO-d6) δ: 180.4, 151.0, 140.3, 134.5, 130.5, 128.7, 128.3, 124.6, 124.3, 119.0, 116.6, 111.1, 95.8, 42.3, 36.8, 33.9, 28.4; HRMS (ESI) m/z: 402.1999 [M+H]+, Calt: 402.1998 [M+H]+.
7d: white solid, yield 78.8%, mp 225~226℃; 1H NMR (400 MHz, DMSO-d6) δ: 10.91 (s, 1H), 9.54 (s, 1H), 8.87 (brs, 1H), 7.42 (d, J=1.5 Hz, 1H), 7.30-7.24 (m, 2H), 7.22 (dd, J=1.5, 6.9 Hz, 1H), 7.19-7.09 (m, 2H), 6.98 (dd, J=1.9, 8.5 Hz, 1H), 6.10 (d, J=1.7 Hz, 1H), 2.24 (s, 3H), 2.07 (brs, 3H), 1.97 (d, J=2.0 Hz, 6H), 1.82 1.72 (m, 6H); 13C NMR (100 MHz, DMSO-d6) δ: 181.1, 151.0, 138.8, 135.3, 134.6, 130.6, 128.7, 128.3, 126.6, 126.3, 119.2, 116.9, 111.2, 95.8, 42.3, 36.8, 33.9, 28.4, 18.4;HRMS (ESI) m/z: 416.2152 [M+H]+,Calt: 416.2155 [M+H]+.
7e: white solid, yield 75.6%, mp 234~236℃;1H NMR (400 MHz, DMSO-d6) δ: 10.91 (s, 1H), 9.62 (s, 1H), 9.27 (brs, 1H), 7.41 (s, 1H), 7.32-7.24 (m, 3H), 7.19 (t, J=8.1 Hz, 1H), 6.97 (dd, J=1.8, 8.5 Hz, 1H), 6.92 (d, J=7.5 Hz, 1H), 6.09 (d, J=1.5 Hz, 1H), 2.28 (s, 3H), 2.07 (brs, 3H), 1.97 (brs, 6H), 1.83-1.63 (m, 6H);13C NMR (100 MHz, DMSO-d6) δ: 180.3, 151.0, 140.2, 137.9, 134.5, 130.5, 128.5, 128.2, 125.3, 124.9, 121.5, 119.1, 116.6, 111.1, 95.8, 42.3, 36.8, 33.9, 28.4, 21.5; HRMS (ESI) m/z: 416.2150 [M+H]+,Calt: 416.2155 [M+H]+.
7f: white solid, yield 79.5%, mp 227~228℃; 1H NMR (600 MHz, DMSO-d6) δ: 10.92 (brs, 1H), 9.59 (brs, 1H), 9.26 (brs, 1H), 7.41 (s, 1H), 7.35 (d, J=8.0 Hz, 2H), 7.27 (d, J=8.4 Hz, 1H), 7.11 (d, J=8.0 Hz, 2H), 6.97 (d, J=7.7 Hz, 1H), 6.09 (s, 1H), 2.28 (s, 3H), 2.07 (brs, 3H), 1.97 (brs, 6H), 1.81-1.70 (m, 6H); 13C NMR (150 MHz, DMSO-d6) δ: 180.4, 151.0, 137.7, 134.5, 133.8, 130.5, 129.1, 128.2, 124.6, 119.1, 116.6, 111.1, 95.7, 42.3, 36.8, 33.9, 28.4, 21.0;HRMS (ESI) m/z: 416.2151 [M+H]+,Calt: 416.2155 [M+H]+.
7g: white solid, yields 80.1%, mp 244~246℃; 1H NMR (400 MHz, DMSO-d6) δ: 10.90 (s, 1H), 9.50 (s, 1H), 9.14 (brs, 1H), 7.41 (d, J=0.9 Hz, 1H), 7.33 (d, J=8.9 Hz, 2H), 7.27 (d, J=8.4 Hz, 1H), 6.97 (dd, J=1.8, 8.5 Hz, 1H), 6.88 (d, J=8.9 Hz, 2H), 6.09 (d, J=1.5 Hz, 1H), 3.74 (s, 3H), 2.07 (brs, 3H), 1.97 (brs, 6H), 1.82-1.71 (m, 6H);13C NMR (100 MHz, DMSO-d6) δ: 180.7, 156.8, 151.0, 134.5, 133.1, 130.5, 128.3, 126.7, 119.1, 116.7, 113.9, 111.1, 95.7, 55.7, 42.3, 36.8, 33.9, 28.4;HRMS (ESI) m/z: 432.2106 [M+H]+,Calt: 432.2104 [M+H]+.
7h: white solid, yields 73.1%, mp 208~210℃; 1H NMR (600 MHz, DMSO-d6) δ: 10.93 (s, 1H), 9.70 (brs, 1H), 9.31 (brs, 1H), 7.46 (dd, J=5.1, 8.6 Hz, 2H), 7.40 (s, 1H), 7.27 (d, J=8.6 Hz, 1H), 7.14 (t, J=8.8 Hz, 2H), 6.96 (dd, J=1.4, 8.5 Hz, 1H), 6.09 (d, J=1.5 Hz, 1H), 2.07 (brs, 3H), 1.97 (brs, 6H), 1.80-1.72 (m, 6H); 13C NMR (150 MHz, DMSO-d6) δ: 180.7, 159.5 (d, J=238.5 Hz), 151.0, 136.7, 134.6, 130.3, 128.3, 126.9, 119.0, 116.6, 115.2(d, J=21.0 Hz), 111.2, 95.8, 42.2, 36.8, 33.9, 28.4; (ESI) m/z: 420.1904 [M+H]+,Calt: 420.1904 [M+H]+.
7i: white solid, yields 79.1%, mp 245~246℃; 1H NMR (600 MHz, DMSO-d6) δ: 10.94 (brs, 1H), 9.78 (brs, 1H), 9.44 (brs, 1H), 7.52 (d, J=8.4 Hz, 2H), 7.41 (brs, 1H), 7.35 (d, J=8.8 Hz, 2H), 7.27 (d, J=8.4 Hz, 1H), 6.96 (d, J=8.0 Hz, 1H), 6.09 (s, 1H), 2.07 (brs, 3H), 1.97 (brs, 6H), 1.81-1.72 (m, 6H); 13C NMR (150 MHz, DMSO d6) δ: 180.4, 151.1, 139.4, 134.6, 128.7, 128.4, 128.2, 126.0, 125.8, 119.0, 116.6, 111.2, 95.8, 42.2, 36.8, 33.9, 28.4; (ESI) m/z: 436.1605 [M+H]+,Calt: 436.1609 [M+H]+.
7j: white solid, yield 82.3%, mp 218~220℃; 1H NMR (400 MHz, DMSO-d6) δ: 10.93 (s, 1H), 9.95 (s, 1H), 9.71 (brs, 1H), 7.78 (d, J=8.4Hz, 2H), 7.65 (d, J=8.6 Hz, 2H), 7.43 (s, 1H), 7.28 (d, J=8.4 Hz, 1H), 6.99 (dd, J=1.7, 8.5 Hz, 1H), 6.10 (d, J=1.5Hz, 1H), 2.07 (brs, 3H), 1.97 (d, J=2.0 Hz, 6H), 1.82-1.69 (m, 6H);13C NMR (100 MHz, DMSO-d6) δ: 180.3, 151.1, 144.3, 134.6, 130.3, 128.3, 125.7, 123.5, 118.8, 116.5, 111.2, 95.8, 42.2, 36.8, 33.9, 28.4; (ESI) m/z: 470.1872 [M+H]+,Calt: 470.1872 [M+H]+.
7k: white solid, yield: 88.6%; mp 242-243℃; 1H NMR (600 MHz, DMSO-d6) δ: 10.78-10.94 (m, 1H), 9.42 (brs, 1H), 8.77 (brs, 1H), 7.40 (s, 1H), 7.26 (d, J=8.4 Hz, 1H), 7.10 (d, J=7.8 Hz, 1H), 7.01 (s, 1H), 6.98-6.92 (m, 2H), 6.14-5.94 (m, 1H), 2.25 (s, 3H), 2.19 (s, 3H), 2.05 (brs, 3H), 1.96 (brs, 6H), 1.82-1.70 (m, 6H);13C NMR (150 MHz, DMSO-d6) δ: 151.0, 135.8, 135.1, 134.6, 131.2, 129.4, 128.7, 128.6, 128.3, 126.9, 119.2, 116.9, 95.8, 42.3, 36.8, 33.9, 28.4, 21.0, 18.3;(ESI) m/z: 430.2310 [M+H]+,Calt: 430.2311 [M+H]+.
7l: white solid, yield 72.8%, mp 228~230℃;1H NMR (400 MHz, DMSO-d6) δ: 10.94 (s, 1H), 9.85 (brs, 1H), 8.96 (brs, 1H), 7.52 (d, J=6.7 Hz, 1H), 7.42 (s, 1H), 7.31-7.23 (m, 2H), 7.07-7.01 (m, 1H), 6.97 (dd, J=1.8, 8.5Hz, 1H), 6.11 (d, J=1.6 Hz, 1H), 2.07 (brs, 3H), 1.97 (d, J=2.2 Hz, 6H), 1.83-1.69 (m, 6H);13C NMR (100 MHz, DMSO-d6) δ: 181.6, 161.7, 156.2, 151.1 (d, J=25.0 Hz), 134.7, 131.0, 130.2 (d, J=25.0 Hz), 129.4, 128.7, 128.3, 124.9, 119.0, 116.8, 111.3, 111.1, 104.5, 95.8, 42.2, 36.8, 33.9, 28.4; (ESI) m/z: 438.1812 [M+H]+,Calt: 438.1810 [M+H]+.
Results and Discussion
Structure-Activity Relationship (SAR) Discussion
The target compounds were synthesized starting from adamantane-1-carbonyl chloride and o-toluidine to yield N-(o-tolyl)adamantane-1-carboxamide (compound 2), which was then reacted with n-butyllithium to give 2-(adamantan-1-yl)-1H-indole (compound 3). Nitration of 3 afforded 2-(adamantan-1-yl)-5-nitro-1H-indole (compound 4), which was reduced to 5-amino-2-(adamantan-1-yl)-1H-indole (compound 5). Treatment of 5 with triphosgene yielded 5-isothiocyanato-2 (adamantan-1-yl)-1H-indole (compound 6). Reaction of 6 with various amines gave the target compounds 7a–7l, whose structures were confirmed by 1H NMR, 13C NMR, and HRMS. The in vitro inhibitory activities of the synthesized compounds against the gastric cancer cell line SGC-7901 were evaluated using the MTT assay, and the results are summarized in Table 1. The effects of different R groups (alkyl, cycloalkyl, substituted aryl, and fused aromatic rings) on the inhibitory activity against SGC-7901 were investigated. According to Table 1, the order of potency for R=hexyl, cyclohexyl, phenyl, and naphthyl was 7c (phenyl) > 7b (cyclohexyl) > 7a (hexyl). For monosubstituted phenyl derivatives (7d–7j), the order was 4-Cl (7i) > 4-F (7h) > 4-OCH3 (7g) > 4-CF3 (7j) > 2-CH3 (7d) > 3-CH3 (7e) > 4-CH3 (7f). For disubstituted phenyl derivatives (7k–7l), 2,4-diCl (7l) > 2,4-diCH3 (7k). Compound 7l exhibited the most potent activity, with an IC50 value of 2.26 ± 0.04 μM. Moreover, 7l showed low toxicity toward normal gastric epithelial cells (IC50>100 μM).
Table 1: Structure, kinase and cell inhibitory activity of the targeted compounds.
Analysis of the Binding Mode of 7l with CDK9
Molecular docking studies were performed to predict the binding mode of 7l with Compound Structure SGC-7901 (IC50/μM) CDK9 inhibiton (@ 1μM) R 7a 15.35 ± 0.23 50.1% 7c 10.11 ± 0.12 58.1% 7e 14.56 ± 0.20 52.3% 7g O 8.45 ± 0.07 73.8% 7i Cl 7.35 ± 0.11 79.5% 7k 13.18 ± 0.47 54.1% 7m 24.16 ± 0.14 45.9% 7b 21.24 ± 0.34 40.2% 7d 12.10 ± 0.45 54.3% 7f 17.23 ± 0.11 49.7% 7h F 8.09 ± 0.37 75.5% 7j CF3 9.76 ± 0.20 60.1% 7l F F 2.26 ± 0.04 85.6% CDK9. The results indicated that 7l binds within the ATP-binding pocket of CDK9, and its three-dimensional structure fits well into the active site (Figure 2A). As shown in Figure 2B, the indole core and thiourea moiety of 7l form stable interactions with key residues of CDK9. The hydrogen atom on the indole nitrogen acts as a hydrogen bond donor, forming a strong hydrogen bond (1.80 Å) with the carbonyl oxygen of GLU107 in the hinge region, indicating that the core of 7l is well-anchored in the hinge region of the CDK9 active site. Additionally, the hydrogen atoms on the thiourea moiety provide further opportunities for interactions with amino acid residues in the active site. The docking results showed that a hydrogen atom on the thiourea group acts as a hydrogen bond donor, forming a hydrogen bond (2.20 Å) with GLY28 of CDK9. Furthermore, the binding free energy of 7l with CDK9, calculated using the MM-GBSA method, was –32.86 kcal/mol, further confirming the high binding affinity of 7l for CDK9.
Through a molecular hybridization strategy, twelve new 1-(2-(adamantan-1-yl) 1H-indol-5-yl)-3-substituted thiourea derivatives (7a–7l) were synthesized and characterized by 1H NMR, 13C NMR, and HRMS. The MTT and CDK9 inhibition assay demonstrated that these compounds inhibit the growth of gastric cancer cells, with compound 7l exhibiting the most potent activity. Molecular docking studies indicated that 7l binds stably to the active site of CDK9 with high binding affinity. Future work will focus on further evaluation of 7l.
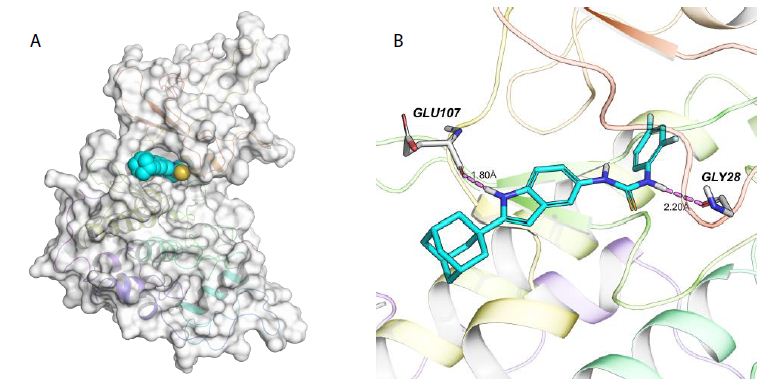
Figure 2: Binding pattern of compound 7l to CDK9. A. Schematic representation of the surface of 7l binding to CDK9; B. The three-dimensional interaction diagram of 7l binding to the protein.
References
- Yan X, Song X, Wang Z (2017) Construction of specific magnetic resonance imaging/optical dual-modality molecular probe used for imaging angiogenesis of gastric cancer. Artificial Cells, Nanomedicine, and Biotechnology 45: 399-403. [crossref]
- Musavi shenas SMH, Mansoori B, Mohammadi A, et al. (2017) SiRNA-mediated silencing of Snail-1 induces apoptosis and alters micro RNA expression in human urinary bladder cancer cell line. Artificial Cells, Nanomedicine, and Biotechnology 45: 969-974. [crossref]
- Liang SH, Yan XZ, Wang BL, Shima S, Behzad K, et al. (2013) Increased expression of FOXQ1 is a prognostic marker for patients with gastric cancer. Tumor Biol 34: 2605-2609. [crossref]
- Shaw RJ, Cantley LC. Ras (2006) PI(3)K and mTOR signalling controls tumour cell growth. Nature 441: 424-430.
- Shibue T, Weinberg RA (2017) EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol 14: 611-629. [crossref]
- Malumbres M (2014) Cyclin-dependent kinases. Genome Biology 15: 1-10.
- Malumbres M, Barbacid M (2005) Mammalian cyclin-dependent kinases. Trends in Biochemical Sciences 30: 630-641. [crossref]
- Loyer P, Trembley JH, Katona R, Vincent JK, Jill ML (2005) Role of CDK/cyclin complexes in transcription and RNA splicing. Cell Signal 17: 1033-1051. [crossref]
- Konecny GE (2016) Cyclin-dependent kinase pathways as targets for women’s cancer treatment. Current Opinion in Obstetrics and Gynecology 28: 42-48.
- Wang X, Gao Y, Li Y, Yuqing H, Yawen Z, et al. (2020) Roseotoxin B alleviates cholestatic liver fibrosis through inhibiting PDGF-B/PDGFR-β pathway in hepatic stellate cells. Cell Death & Disease 11.
- KOHOUTEK J (2009) P-TEFb- the final frontier. Cell Division 4: 1-15. [crossref]
- Wang S, Fischer PM (2008) Cyclin-dependent kinase 9: a key transcriptional regulator and potential drug target in oncology, virology and cardiology. Trends in Pharmacological Sciences 29: 302-313. [crossref]
- Whittaker SR, Barlow C, Martin MP, Caterina M,, Steve W, et al. (2018) Molecular profiling and combinatorial activity of CCT068127: a potent CDK2 and CDK9 inhibitor. Molecular Oncology 12: 287-304. [crossref]
- Brägelmann J, Dammert MA, Dietlein F, Johannes MH, Axel C, et al. (2017) Systematic Kinase Inhibitor Profiling Identifies CDK9 as a Synthetic Lethal Target in NUT Midline Carcinoma. Cell Reports 20: 2833-2845. [crossref]
- Rahaman MH, Kumarasiri M, Mekonnen LB, Mingfeng Y, Sarah D, et al. (2016) Targeting CDK9: a promising therapeutic opportunity in prostate cancer. Endocrine-Related Cancer 23: T211- T226. [crossref]
- Mitra P, Yang RM, Sutton J, Robert GR, Thomas JG (2016) CDK9 inhibitors selectively target estrogen receptor positive breast cancer cells through combined inhibition of MYB and MCL-1 expression. Oncotarget 7: 9069-9083. [crossref]
- Baker A, Gregory GP, Verbrugge I, Lev K, Joshua JH, et al. (2016) The CDK9 Inhibitor Dinaciclib Exerts Potent Apoptotic and Antitumor Effects in Preclinical Models of MLL-Rearranged Acute Myeloid Leukemia. Cancer Research 76: 1158-1169. [crossref]
- Ajiro M, Sakai H, Onogi H, Makoto Yamamoto, et al. (2018) CDK9 Inhibitor FIT-039 Suppresses Viral Oncogenes E6 and E7 and Has a Therapeutic Effect on HPV-Induced Neoplasia. Clinical Cancer Research 24: 4518-4528. [crossref]
- Lu Y, Tang L, Zhang Q, et al. (2018) MicroRNA-613 inhibits the progression of gastric cancer by targeting CDK9. Artificial cells, nanomedicine, and biotechnology 46: 980-984. [crossref]