Abstract
Treacher Collins syndrome (TCS) is a rare congenital craniofacial dysplasia characterized by malformations of the jaw, eyes, and ears, with an incidence of approximately 1 in 50000.This study reported a case of a 4-year-old male TCS patient presenting with cleft palate. The patient exhibited facial asymmetry, flat zygomatic bone and zygomatic arch, mandibular hypoplasia, and eye and ear abnormalities. Genetic testing confirmed a POLR1 D gene mutation, leading to a diagnosis of TCS type II. The patient underwent cleft palate repair surgery and demonstrated significant recovery postoperatively. Follow-up evaluations showed significant improvement in speech and palatal function. The diagnosis of TCS relies on clinical manifestations, imaging examinations, and genetic testing. Effective treatment necessitates multidisciplinary collaboration, encompassing craniofacial reconstruction, hearing enhancement, and speech therapy. A comprehensive treatment plan should be tailored to the patient’s age and severity of deformities to address the physiological function and psychological needs. Future efforts should focus on enhancing the application of molecular genetics in the diagnosis and treatment of TCS to improve prenatal diagnostic capabilities.
Keywords
Treacher Collins syndrome, Cleft palate
Introduction
Treacher Collins syndrome (TCS) is a rare congenital craniofacial dysplasia characterized by malformations of the jaw, eyes, and ears, with an incidence of approximately 1 in 50000 [1]. First reported by Edward Treacher Collins in 1900 [2], the syndrome can be classified into four clinical subtypes. Its primary clinical features include blepharophimosis, zygomatic dysplasia, conductive deafness, and mandibular dysplasia or micrognathia, with cleft palate cases being particularly rare [3,4]. This paper presents a case of a TCS patient with cleft palate, detailing the symptoms, diagnosis, treatment process, and follow-up results to provide a reference for clinicians in the diagnosis and treatment of this condition.
Case Report
A 4-year-old male patient was identified with facial deformity and cleft palate at birth. Examination revealed facial asymmetric, flat bilateral zygoma and zygomatic arch, a short mandibular ramus, wide orbital spacing, oblique palpebral fissures, absence of the lower eyelid, and missing lower eyelid eyelashes (Figure 1). The patient also exhibited bilateral auricle deformity, external auditory canal atresia with preauricular fistula (Figure 2), and a cleft palate extending from the uvula to the incisive foramen, with a maximum width of 2.5 centimeters (Figure 3). Hearing tests indicated moderate conductive hearing loss in both ears. Genetic analysis identified a heterozygous spontaneous mutation in the POLR1 D gene, leading to a diagnosis of TCS type II. The patient’s speech was unclear due to the cleft palate. Upon admission, the patient underwent cleft palate repair surgery under general anesthesia with tracheal intubation. Postoperative recovery was successful (Figure 4), and voice training commenced three months post-surgery. Follow-up evaluations at 6 months, 12 months, and 24 months post-operation showed well-recovered palate morphology, normal soft palate movement, significantly improved pronunciation, and no coughing during meals.
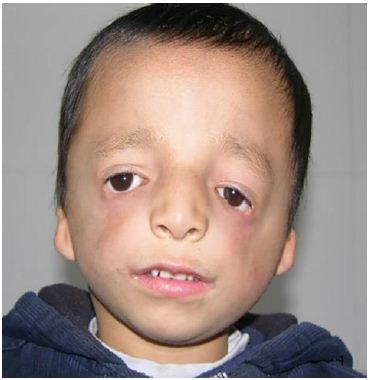
Figure 1: Facial asymmetric, flat bilateral zygoma and zygomatic arch, a short mandibular ramus, wide orbital spacing, oblique palpebral fissures, absence of the lower eyelid, and missing lower eyelid eyelashes.

Figure 2: Bilateral auricle deformity, external auditory canal atresia with preauricular fistula.
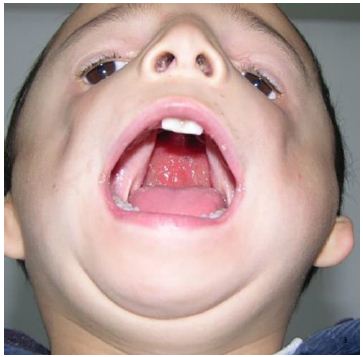
Figure 3: A cleft palate extending from the uvula to the incisive foramen, with a maximum width of 2.5 cm.
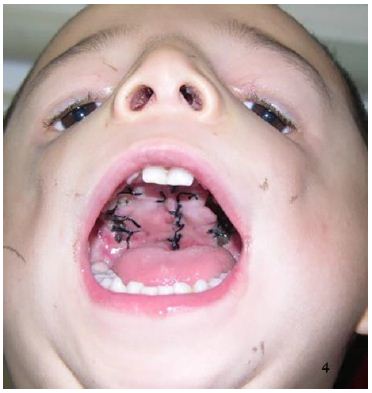
Figure 4: The palate healed well
Discussion
TCS is a rare craniofacial malformation with autosomal dominant inheritance, also known as maxillofacial dysplasia and deafness syndrome. It presents a highly variable phenotype, with about 40% of patients having a family history [3]. The cause of TCS is attributed to disrupted ribosome synthesis in cranial neural crest cells and neuroepithelial cells between the 2nd and 8th weeks of embryonic development. This disruption leads to a reduction in the number of neural crest cells migrating to the craniofacial region, resulting in hypoplasia of the first and second branchial arches [5].
TCS can present with various clinical types. According to studies by Splendor (2000), Teber (2004), and Vincent [6-8], the clinical features of TCS patients can be summarized as follows: (1) Craniofacial abnormalities: These include facial asymmetry, a low hairline, and facial hypoplasia, particularly affecting the mandibular and zygomatic complex. (2) Eye abnormalities: These are characterized by oblique palpebral fissures and lower eyelid defects. (3) Ear abnormalities: These include atresia of the external auditory canal, microtia, and conductive hearing loss, contributing to a characteristic ‘fish-like ‘ facial appearance. (4) Other rare clinical features: Dental abnormalities such as missing teeth (tooth dysplasia), tooth discoloration (enamel opacity), excessive tooth spacing, abnormal permanent teeth (eg, ectopic maxillary first molars), and occlusal disorders. Palatal abnormalities such as high-arched palate and cleft palate. Respiratory and feeding difficulties may arise from posterior nostril stenosis or atresia. Cardiac malformations are also possible in some cases [9,10]. Patients with a mild phenotype of TCS may exhibit almost no obvious clinical features and may require genetic testing for identification. Conversely, those with a severe phenotype may experience life-threatening ventilatory disorders due to obstruction of the posterior nasal foramen, glossoptosis, and other complications [11].
The clinical diagnosis of TCS is primarily based on clinical manifestations, imaging examinations, and pathogenic gene detection. X-ray imaging typically reveals several characteristic features: increased density of small mastoid bones, nasal protrusion with a wide, flat frontonasal angle, maldevelopment or defects in the zygomatic bone and zygomatic arch, narrow maxillary protrusion, small maxillary sinus, mandibular dysplasia with a short body and ascending ramus, and a deepened anterior corner notch. Ultrasound examination is valuable for the intrauterine diagnosis of TCS, with the fetus often presenting with polyhydramnios, absence of fetal swallowing activity, and poor development of the bilateral parietal diameter and head circumference. Known pathogenic genes associated with TCS include TCOF1, POLR1C, and POLR1D. Mutations in these genes can lead to reduced ribosomal RNA transcription and ribosome synthesis, which subsequently affect the development and differentiation of neural crest cells during the embryonic stage [6,7,12].
The clinical manifestations of TCS are similar to those of several other syndromes, necessitating differential diagnosis. These syndromes include Nager syndrome, Miller syndrome, Goldenhar syndrome, and Pierre Robin syndrome [13,14]. The facial features of Nager syndrome are similar to those of TCS, but Nager syndrome also presents with typical limb deformities such as thumb dysplasia or absence, polydactyly, and radial ulna bony fusion [15]. Miller syndrome is characterized by asymmetric upper and lower eyelid ectropion and defects, dysplasia of the fifth finger (toe), and a higher prevalence of cleft lip and palate compared to TCS [16]. Goldenhar syndrome is marked by hemifacial atrophy affecting the development of the ears, mouth, and mandible, and may also include vertebral abnormalities and dermoid cysts on the outer layer of the eye [14]. Pierre Robin syndrome is distinguished by micrognathia, glossoptosis, dyspnea, and cleft palate [17].
The comprehensive sequential treatment of TCS typically begins at birth and continues until growth and development are complete. Treatment should be tailored to the patient’s growth pattern, physiological function, and psychosocial needs. It is recommended to ensure and maintain basic life functions before the age of 2 years. If there is persistent corneal exposure between the ages of 2 and 5 years, orbital wall reconstruction should be performed for correction. If the mandible is underdeveloped between the ages of 6 and 10 years, mandibular traction should be performed. Speech therapy, craniofacial fracture reconstruction, and external ear reconstruction should be completed before the age of 12. Orthodontic-orthognathic treatment, correction of upper and lower jaw and nasal deformities, and social psychotherapy should be carried out between the ages of 13 and 18 years. Future treatment should incorporate molecular genetics, utilizing genetic tests for prenatal examinations in high-risk groups and monitoring fetal growth and development during the first three months of pregnancy.
The phenotypes of TCS vary greatly, ranging from mild to severe deformities. For example, airway obstruction caused by mandibular deformity can affect breathing, and eyelid defects can expose the cornea. Therefore, TCS treatment should follow a multidisciplinary comprehensive sequence approach, involving oral and maxillofacial surgery, plastic surgery, orthodontics, ophthalmology, otolaryngology, speech therapy, psychology, genetics, and nursing. Individualized treatment measures should be selected based on the patient’s age and degree of deformity.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Statement
The studies involving human participants were reviewed and approved by the ethics committee of Qingdao Women and Children’s Hospital
Informed Consent
The patient ‘s legal guardian provided written informed consent to participate in this study.
Author’s Contribution
Ting li: Conception and design of study, Acquisition of data, Data analysis and interpretation, Drafting of manuscript and critical revision, Approval of final version of manuscript.
Yuelin qin: Conception and design of study, Acquisition of data, Data analysis and interpretation, Drafting of manuscript and critical revision, Approval of final version of manuscript.
Ziyan lu: Acquisition of data.
Xuecai yang: Drafting of manuscript and critical revision, Approval of final version of manuscript.
Junwei wang: Drafting of manuscript and critical revision, Approval of final version of manuscript.
References
- Nguyen PD, Caro MC, Smith DM, et al. (2016) Long-term orthognathic surgical outcomes in Treacher Collins patients. J Plast Reconstr Aesthet Surg 69: 402-408. [crossref]
- Treacher Collins E (1900) Case with symmetrical congenital notches in the outer part of each lower lid and defective development of the malar bones. Trans Opthalmol Soc 20: 190-192.
- Shinde CV, Kohli S (2018) Treacher Collin’s syndrome: a case report with an augmented review! International Journal of Advanced Research 6: 141-146.
- Renju R, Varma BR, Kumar SJ, et al. (2014) Mandibulofacial dysostosis (Treacher Collins syndrome): A case report and review of literature. Contemporary Clinical Dentistry 5: 532-534.
- van Gijn DR, Tucker AS, Cobourne MT (2013) Craniofacial development: current concepts in the molecular basis of Treacher Collins syndrome. Br J Oral Maxillofac Surg 51: 384-388.
- Splendore A, Silva EO, Alonso LG, et al. (2000) High mutation detection rate in TCOF1 among Treacher Collins syndrome patients reveals clustering of mutations and 16 novel pathogenic changes. Hum Mutat 16: 315-322. [crossref]
- Teber OA, Gillessen-Kaesbach G, Fischer S, et al. (2004) Genotyping in 46 patients with tentative diagnosis of Treacher Collins syndrome revealed unexpected phenotypic variation. Eur J Hum Genet 12: 879-890. [crossref]
- Vincent M, Geneviève D, Ostertag A, et al. (2016) Treacher Collins syndrome: a clinical and molecular study based on a large series of patients. Genet Med 18: 49-56. [crossref]
- Campos PSSL, Taitson PF, Pinto da Silva LC, et al. (2022) Dental and health aspects in the co-occurrence of Treacher Collins and Down syndromes: Case report. Spec Care Dentist 43: 94-98. [crossref]
- Kadakia S, Helman SN, Badhey AK, et al. (2014) Treacher Collins syndrome: the genetics of a craniofacial disease. Int J Pediatr Otorhinolaryngol 78: 893-898. [crossref]
- Islam F, Afroza A, Rukunuzzaman M, et al. (2008) Treacher Collins Syndrome-A Case Report. Bangladesh Journal of Child Health 32: 33-36.
- Bowman M, Oldridge M, Archer C, et al. (2012) Gross deletions in TCOF1 are a cause of Treacher–Collins-Franceschetti syndrome. European Journal of Human Genetics 20: 769-777. [crossref]
- Thomas P, Krishnapillai R, et al. (2019) Treacher Collins Syndrome: A Case Report and Review of Literature. Oral Maxillofac Patho J 10: 90-94.
- Shete P, Tupkari J, Benjamin T, et al. (2011) Treacher Collins syndrome. Journal of Oral and Maxillofacial Pathology 15: 348-351.
- Rosa RF, Guimarães VB, Beltrão LA, et al. (2015) Nager syndrome and Pierre Robin sequence. Pediatrics international: official journal of the Japan Pediatric Society 57: e69-72. [crossref]
- Trainor PA, Andrews BT (2014) Facial dysostoses: Etiology, pathogenesis and management. American Journal of Medical Genetics Part C Seminars in Medical Genetics 163: 283-294.
- Shen YF, Vargervik K, Oberoi S, et al. (2012) Facial Skeletal Morphology in Growing Children With Pierre Robin Sequence. Cleft Palate Craniofac J 49: 553-560.