DOI: 10.31038/CST.2017273
Short Communication
The etiology of AD and other age-related neurodegenerative disorders is complex, involving as it does, many multivariate interacting pathways among which are aging itself, inflammation, mitochondrial ROS disorders, hyperactive kinases such as Cdk5, mutations in amyloid precursor protein (APP), secretases and tau processing. The identification of gene mutations responsible for neurodegeneration in humans has led to development of a variety of transgenic mouse models, each expressing a neurodegenerative phenotype such as AD, PD or ALS. For AD, for example, an extensive model mouse literature has accumulated validating one or another pathway as specifically responsible for the behavioral and pathological disease phenotype (e.g. Elder et al, 2010, Landreth et al, 2012). And, in some instances, these mutant mice have been claimed successfully “cured” or “rescued” to some extent by agents or manipulations that restore the damaged pathways. These are touted as potential therapeutic candidates.
Our current research program evolved after years of basic science studies of neuronal cytoskeletal protein phosphorylation during nervous system development and function, A brief summary of our accomplishments over the years should illustrate how we arrived at our present program. We found that neurofilaments, the major axonal proteins, are selectively phosphorylated in axons [1-4]. Using a neurofilament assay, our laboratory identified cyclin dependent kinase 5 (Cdk5), together with its activator, P35, as one of the principal kinases regulating neuronal topographic phosphorylation biology and physiology. The multifunctional kinase, Cdk5, was initially characterized as a tau protein kinase (Ishiguro et al., 1991), a proline directed kinase (Lew et al., 1994; Lew et al., 1995; Lew and Wang, 1995) or a cdc-2 like kinase in our lab (Shetty et al 1993, Shetty et al., 1995), presently known as Cdk5. We as well as other laboratories have shown that Cdk5 is a tightly regulated multifunctional kinase essential for neuronal development, neurogenesis, migration, synaptic activity, memory / learning and survival, phosphorylating a large number of target protein substrates [5-10].
Cdk5 when deregulated by neuronal stress (e.g., glutamate excitotoxicity, A toxicity, Reactive Oxygen Species ,ROS, and others), Cdk5 activity is deregulated and hyperactivated as a stable complex with p25 (a truncated fragment of p35, a major activator of Cdk5) and induces perikaryal hyperphosphorylated tau, neurofilament proteins (NF-M/H), and other neuronal intermediate filament proteins as seen in AD, PD and ALS [11, 12], thus Cdk5/p25 becomes a pathological target. This relationship to AD and other pathologies has been documented in studies of AD brains showing high levels of p25, reduced p35/p25 ratios and Cdk5 hyperactivity [13, 14] Zheng et al 2010, Furthermore, we have confirmed that Cdk5/p25 induces tau and NFP aberrant hyperphosphorylation along with cell death in cultured cortical neurons [14,15]. Consistent with this hypothesis is a p25-overexpressing model mouse, developed by Dr. Tsai’s lab and recently our own lab, that displays the typical AD abnormal phenotype [16]. Accordingly, hyperactive Cdk5/p25 has been identified as a possible therapeutic target for neurodegeneration. Recently, a more compelling role of hyperactive Cdk5/p25 as a significant factor in the etiology of AD comes from studies of cell cultures in vitro [14,15] and model mice in vivo as documented in two publications. Since the binding of p25, the proteolytic fragment of p35, induces deregulated and hyperactivated Cdk5, we asked the question what is the role of smaller truncated peptides of p25 in the regulation of Cdk5 activity? This has led us to isolation and identification of peptides derived from p35 (CIP , cdk5 inhibitory peptide and P5, a 24 amino acid peptide derived from p25) that specifically inhibited the hyperactive Cdk5/p25 without affecting the physiologically normal Cdk5/p35 [12,14,15] (Amin et al., 2002] Consistent with the model, we succeeded in showing that pathological and behavioral phenotypes in AD model mice (over-expressing p25 transgenic) and the 5XFAD transgenic can be alleviated after treatment with CIP and TFP5, our in vivo therapeutic reagents (Sundaram et al, 2013, Shukla et al, 2013).
We viewed these peptides as potential therapeutic candidates for rescuing neurodegenerative disorders in model mice sharing the hyperactivated Cdk5-induced phenotypes. Currently, most therapeutic approaches targeting the deregulated Cdk5/p25 complex and other kinases in neurodegenerative disorders and cancer have focused primarily on drugs like roscovitine, ATP analog, that inhibit Cdk5 activity by interfering with the ATP binding domain of the kinase. Most of these drugs, however, lack sufficient specificity, since all kinases including cell cycle Cdks including Cdk5, are vulnerable at the ATP binding site targeted by these drugs. In order to make P5, small 24 amino acid peptide an in vivo therapeutic reagent, we coupled the C-terminus of P5 to a protein transduction domain peptide (PTD -TAT) and its N-terminus to FITC, fluorescent tag, fluorescein isothiocynate. This reagent that we call TFP5 was shown to pass the blood brain barrier and to rescue the AD phenotype in AD model mice (Shukla et al, 2013, Sudaram et al, 2013). Hence, next, we conducted the following studies:
Effects of TFP5 on expression of AD phenotypes in a p25Tg over-expressing and double transgenic (5XFADTg) AD model mice, mechanisms of specificity of TFP5 action and effect of TFP5 on ALS model mice (unpublished).
At the forefront of the AD literature are those model mice expressing the two landmark pathologies seen at human autopsy, hyper phosphorylated intracellular cytoskeletal tangles (tau, neurofilaments) and extracellular amyloid plaques. One of the many hypotheses invoked to explain these phenotypes is the role of deregulated, hyperactive Cdk5/p25, reported at elevated levels in AD brains at autopsy (Ishiguro et al, 1991; Ishiguro et al, 1992; Patrick et al, 1999; Tseng et al, 2002). From these and other related studies in vivo and in vitro, a hypothesis has been proposed to account for the deregulation of Cdk5, and its induction of tau and amyloid pathology leading to the chronically long descent into the abyss of dementia. (Figure 1). A most persuasive validation of the hypothesis is overexpression of p25 in a mouse model that induces Cdk5 hyperactivity, AD pathology, behavioral defects and early mortality [16] (Cruz et al, 2003). Elevated levels of p25 in post-mortem AD brains, however, though supported by the above (and other reports), was not confirmed in a few laboratories (Nguyen et al,2002; Tandon et al, 2002; Kerokoski et al, 2002; Takashima et al, 2001). These differences had been, in part, attributed to tissue sampling conditions and preparation protocols [17-21]. Nevertheless, the weight of evidence from our lab and others is consistent with the hypothesis; hyperactive Cdk5/p25 has been identified as a target in neurodegeneration and many compounds that inhibit this kinase have been tested in AD model mice (Glicksman et al, 2007; Hassan et al, 2011; Demange et al, 2013; Shukla et al, 2013; Sundaram et al, 2013).
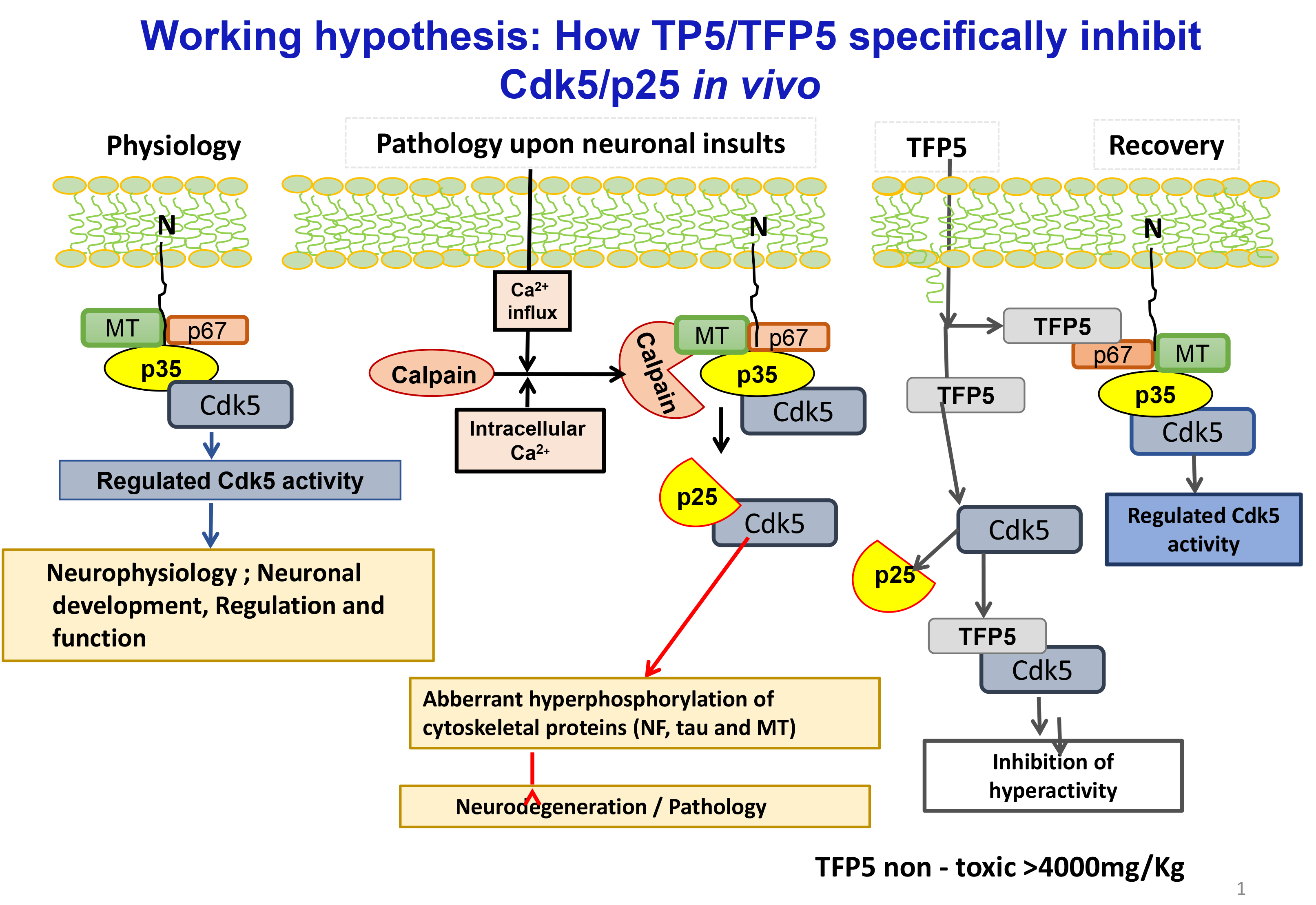
Figure 1. In physiological state N- terminus domain (10kDa) of p35 which anchoring to the membrane, the activator of Cdk5 forms a multimeric complex associated with Cdk5 and a large number of neuronal cytoskeletal and synaptic proteins ( e.g. p67) regulates a large number of neuronal processes , essential for nervous system development and survival ( Fig.1, physiology). Upon neuronal toxic insults/ stress factors (glucose , Aß, oxidative and other)induces intracellular Ca+ increase which activates calcium dependent proteases (e.g., calpain), cleaves p35 into 10kDa N- terminus domain and a 25 kDa truncated protein , which has higher affinity for Cdk5 leads hyperactivates Cdk5 activity. This aberrant and hyperactive Cdk5 ( Cdk5/p25)hyperphosphorylates a large number of cytoskeletal proteins including tau, Neurofilament produces neurodegeneration and pathology. Thus Cdk5/p25 becomes a pathological and Cdk5/p35 physiological target (Fig.1, Pathology). To study the effects of smaller peptides derived from p25 on phosphorylation activity of Cdk5/p35 and Cdk5/p25, p25 polypeptide was truncated to smaller peptides. The smallest peptide a 24 amino acid (TFP5) selectively inhibited deregulated Cdk5 hyperactivity but not cdk5/p35 activity (Fig.1, Recovery)
References
- Link WT, Dosemeci A, Floyd CC, Pant HC (1993) Bovine neurofilament-enriched preparations contain kinase activity similar to casein kinase I–neurofilament phosphorylation by casein kinase I (CKI). Neurosci Lett151: 89–93. [crossref]
- Pant AC, Veeranna, Pant HC, Amin N (1997) Phosphorylation of human high molecular weight neurofilament protein (hNF-H) by neuronal cyclin-dependent kinase 5 (cdk5). Brain Res 765: 259–266. [crossref]
- Pant HC, Veeranna (1995) Neurofilament phosphorylation. Biochem Cell Biol73: 575–592. [crossref]
- Pant HC, Veeranna, Grant P (2000) Regulation of axonal neurofilament phosphorylation. Curr Top Cell Regul36: 133–150. [crossref]
- Chae T, Kwon YT, Bronson R, Dikkes P, Li E, et al. (1997) Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron 18: 29–42. [[crossref]
- Kesavapany S, Li BS, Amin N, Zheng YL, Grant P, et al. (2004) Neuronal cyclin-dependent kinase 5: role in nervous system function and its specific inhibition by the Cdk5 inhibitory peptide. Biochim Biophys Acta1697: 143–153. [crossref]
- Li BS, Zhang L, Takahashi S, Ma W, Jaffe H, et al. (2002) Cyclin-dependent kinase 5 prevents neuronal apoptosis by negative regulation of c-Jun N-terminal kinase 3. EMBO J 21: 324–333. [crossref]
- Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, et al. (1996) Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci U S A93: 11173–11178. [crossref]
- Tanaka T, Veeranna, Ohshima T, Rajan P, Amin ND, et al. (2001) Neuronal cyclin-dependent kinase 5 activity is critical for survival. J Neurosci21: 550–558. [crossref]
- Dhavan R, Tsai LH (2001) A decade of CDK5. Nat Rev Mol Cell Biol2: 749–759. [crossref]
- Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, et al. (2000) Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405: 360–364. [crossref]
- Kesavapany S, Li BS, Pant HC (2003) Cyclin-dependent kinase 5 in neurofilament function and regulation. Neurosignals12: 252–264. [crossref]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, et al. (1999) Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402: 615–622. [crossref]
- Zheng YL, Kesavapany S, Gravell M, Hamilton RS, Schubert M, et al. (2005) A Cdk5 inhibitory peptide reduces tau hyperphosphorylation and apoptosis in neurons. EMBO J 24: 209–220. [crossref]
- Zheng YL, Li BS, Amin ND, Albers W, Pant HC (2002) A peptide derived from cyclin-dependent kinase activator (p35) specifically inhibits Cdk5 activity and phosphorylation of tau protein in transfected cells. Eur J Biochem269: 4427–4434. [crossref]
- Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH (2003) Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron 40: 471–483. [crossref]
- Ahlijanian MK, Barrezueta NX, Williams RD, Jakowski A, Kowsz KP (2000) Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci U S A 97: 2910–5.
- Grant P, Sharma P, Pant HC (2001) Cyclin-dependent protein kinase 5 (Cdk5) and the regulation of neurofilament metabolism. Eur J Biochem 268: 1534–1546. [crossref]
- Lew J, Wang JH (1995) Neuronal cdc2-like kinase. Trends Biochem Sci 20: 33–37. [crossref]
- Shea TB, Yabe JT, Ortiz D, Pimenta A, Loomis P, et al. (2004) Cdk5 regulates axonal transport and phosphorylation of neurofilaments in cultured neurons. J Cell Sci 117: 933–941. [crossref]
- Tsai LH, Delalle I, Caviness VS, Jr., Chae T, Harlow E (1994) p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 371: 419–23.