Abstract
It has generally been assumed that hormones and the corresponding intra- and intercellular signal transduction pathways and mechanisms have evolved exclusively during course of the evolution of vertebrate endocrine organs, implying a rather recent origin. However, there is good experimental evidence for (i) the expression of hormones and hormone-binding proteins resembling those of vertebrates in fungi and yeast, (ii) functional responses of lower eukaryotes to mammalian hormones and (iii) the existence of components of insulin-like and mimetic signaling pathways as well as their coupling to G-protein coupled receptors and metabolic pathways, such as lipolysis and endoplasmic reticulum stress, in lower eukaryotes, in particular in Neurospora crassa and Saccharomyces cerevisiae. Data will be presented that the naturally occurring or recombinant expression of insulin-like/mimetic signaling pathways in lower eukaryotic cells may be useful as model systems for future drug screening and discovery efforts.
Keywords
Diabetes; Drug Screening; Endoplasmic Reticulum Stress; Glucagon-like Peptides; G-protein-coupled Receptors; Insulin; Lipolysis, Saccharomyces cerevisiae
1. Introduction: Models for the discovery of insulin-mimetic and anti-diabetic drugs
The yeast Saccharomyces cerevisiae, as a simple and well-understood eukaryotic model organism, offers direct means to unequivocally assess processes that couple ligand binding to receptor activation, formation of transient intermediates, and downstream signaling to the molecular targets of the activated receptor. For example, events proximal to insulin-binding at the receptor can be traced to distal events to up to the terminal effector systems, such as dephosphorylation of GS or phosphorylation of ribosomal proteins. Furthermore, the interactions between multiple mutations, their suppressors, and ultimately, between the molecular participants, in total, in an insulin-activated response cascade can be readily tested in yeast. The ease of biochemical, cell biological, genetic and molecular biological manipulation has made S. cerevisiae an excellent model for the study of metabolic control in eukaryotic cells. In fact, yeast can be considered as the preferred model eukaryotic organism for the following reasons: I) Many cellular processes are conserved between yeast and higher eukaryotes, including (i) metabolic and energy-generating pathways, (ii) signal transduction pathways, (iii) protein targeting to specific subcellular locations, e.g. secretion, biogenesis of plasma membranes and mitochondria, (iv) peptide maturation, e.g. signal sequence cleavage, prohormone cleavage at Lys-Arg residues, (v) protein modification, e.g. Ser/Thr/Tyr phosphorylation, N- and O-glycosylation, acylation, attachment of GPI anchors, (vi) nuclear processes, e.g. transcription, RNA processing and polyadenylation. II) Many proteins of yeast are very close orthologs to mammalian counterparts, and their functional replacement by the latter has been shown for some yeast components (Table 1), signal transduction components such as protein kinases and phosphatases (Table 2). III) Yeast represents the best understood eukaryotic organism at the genetic level under the control of a compact and simple genome with few introns, repetitive sequences and non-coding elements. IV) Yeast is ideally suited for both classical and molecular genetic manipulation with many powerful tools introduced during the past five decades, including (i) high and low copy number plasmid vectors, (ii) strong and inducible promoters, (iii) high transformation efficiency (> 105/µg of DNA), (iv) gene replacement in one week, (v) identification and cloning of genes acting along a pathway with the methods of functional complementation, genetic suppression, synthetic lethality etc.
Table 1. Structural comparison of some signal transduction proteins/genes between mammals and yeast with regard to %identity of amino acids. As indicated some mammalian genes can restore the function of the disrupted homologous gene in Saccharomyces cerevisiae.
|
Mammalian Protein |
Yeast Gene |
% Identity |
Functional Replacement |
|
Cell Division Protein Kinase |
CDC28 |
60 |
yes |
|
MAP Kinases ERK1, 2 |
FUS3/KSS1 |
54 |
yes |
|
JNK1 |
HOG1 |
52 |
yes |
|
Protein Kinase Cγ |
PKC1 |
50 |
no |
|
Osmotic Stress Kinase p38 |
HOG1 |
52 |
yes |
|
ser/thr Phosphatase PP1 |
PP1 |
81 |
no |
|
p21Ras |
RAS1/RAS2 |
50–85 |
yes |
|
GTP-binding Protein Gp |
CDC42 |
80 |
yes |
|
Gsα |
GPA1 |
40–50 |
yes |
|
G1 Cyclins |
CLN1, 2, 3 |
yes |
|
|
Phospholipase Cγ2 |
PLC1 |
27–53 |
no |
|
Transcription factor Egr1/2 |
Msn4 |
yes |
|
|
Triglyceride Lipase ATGL |
Tgl4 |
yes |
Table 2. Some selected yeast homologs of mammalian protein kinases and phosphatases with established function in signal transduction.
|
Protein Kinase or Phosphatase |
Yeast Homolog |
|
AMP-Activated Protein Kinase (AMPK) |
SNF1 |
|
Calcineurin B (catalytic subunit, Ca2+-dependent serine/threonine phosphatase) |
CNA1, CNA2 (CMP1, CMP2) |
|
Calcineurin B (regulatory subunit, Ca2+-dependent serine/threonine phosphatase) |
CNB |
|
Calmodulin Kinase II |
CMK1, CMK2 |
|
cAMP-Dependent Protein Kinase (catalytic subunit) |
TPK1 (SRA3), TPK2, TPK3 |
|
cAMP-Dependent Protein Kinase (regulatory subunit) |
BCY1 (SRA1) |
|
Cell Cycle Kinase (p34CDC2) |
CDC28 |
|
Cell Division Kinase (CDK3) |
CDC28 |
|
Cyclin Dependent Kinase (CDK2) |
CDC28 |
|
Cytosolic Tyrosine Phosphatase (MEG2) |
SEC14 |
|
Glycogen Synthase Kinase 3 |
MDS1 |
|
Lipopolysaccharide-activated Kinase (JNK1, c-jun amino-terminal protein kinase) |
HOG1 |
|
MAP Kinase Kinase (MKK or MEK) |
STE7, MKK1, MKK2, PBS2 |
|
MAP Kinase Kinase Kinase |
BCK1, STE11 |
|
MAP Kinases (ERK1, ERK2) |
FUS3, KSS1, MPK1 (SLT2), |
|
p65PAK Serine/Threonine Kinase (binds Rac and CDC42) |
STE20 |
|
Protein Kinase C Inhibitor |
BMH1 |
|
Protein Kinase Cb1 |
PKC1 |
|
Serine/Threonine Phosphatase (PP2C) |
PTC1 |
|
Serine/Threonine Phosphatase 2A (B-subunit) |
CDC55 |
|
Tyrosine Phosphatase |
PTP1, PTP2 |
|
Casein Kinase 1 |
HRR25, CKI1, CKI2, CKI3 |
|
Glycogen Synthesis Kinase 3β |
RIM11 |
2. G-Protein Coupled Receptors as Targets
G-protein-coupled receptors (GPCRs) as transmembrane proteins represent components of intracellular signaling cascades which transduce information from the cell surface into the cell interior in course of agonist binding and the resulting activation of heterotrimeric G-proteins. GPCRs together with their corresponding elements of downstream signaling have been found to operate in all eukaryotes from yeast to mammals [1] and to share pronounced sequence similarities between yeast and mammals (Table 3). It has recently been recognized that in mammalian cells a complex interplay and mutual cross-talk exists between the insulin signaling cascade and the signaling pathways directed by GPCRs. It depends on the type of GPCR whether insulin action is modulated in a positive or negative fashion and thus insulin sensitivity becomes increased or diminished (i.e. insulin sensitizing or desensitizing) [2–8]. The basis of the successful screening for anti-diabetic drugs is formed by modeling of the activation of the G-protein-coupled class of human receptors in yeast. The successful coupling of human receptor activation to the mating pathway of yeast has been demonstrated [9–11] which should thereby enable the screening for both agonists and antagonists, depending on the needs. The putative targets encompass known members of the GPCR class which are assumed to be involved in the pathogenesis of T2D, among them the glucagon receptor, glucagon-like-peptide 1 receptor (GLP-1) and a variety of α- and β-adrenergic receptor subtypes, such as muscarinic acetylcholine receptors and various receptors of the brain and nervous system. Significant applications to many other therapeutic areas can be expected where GPCR members play a role.
Table 3. Some selected yeast homologs of mammalian cAMP and G-protein signaling components, acylation factors, and lipid kinases with established function in signal transduction.
|
Signal Transduction Component |
Yeast Homolog |
|
Adenylyl Cyclase-associated Protein (CAP) |
CAP |
|
C-Farnesyl-Cysteine Methyltransferase |
STE14 |
|
Calnexin, Calreticulin |
CNE1 |
|
cAMP-specific Phosphodiesterase (type 3) |
PDE1, PDE2 (SRA5) |
|
Farnesyltransferase, CAAX-specific, Peptide-binding Subunit |
DPR1 (RAM1) |
|
GDP-GTP Exchange Factor |
BUD5, SCD25 |
|
GDP-GTP Exchange Factor (m-SOS, Ras-specific) |
CDC25 |
|
GDP-GTP Exchange Factor (Rab-specific) |
DSS4 |
|
Geranylgeranyl Transferase (Rab-specific) α-subunit |
MAD2 |
|
Geranylgeranyl Transferase (Rab-specific) β–subunit |
BET2 |
|
GTP-Binding Protein (p21Ras) |
RAS1, RAS2 |
|
GTP-Binding Protein (Gas) |
CDC70 (SCG1, GPA1, DAC1) |
|
GTP-Binding Protein (Gb1, Gb2) |
STE4 |
|
GTP-Binding Protein (Ggt) |
STE18 |
|
GTP-Binding Protein, 70% identical to Rac (Gp) |
CDC42 |
|
GTP-Binding Protein, Ras-related (Rab1) |
YPT1 |
|
GTP-Binding Protein, Ras-related (Rab3) |
SEC4, YPT1 |
|
GTP-Binding Protein, Ras-related (Rab7) |
YPT7 |
|
GTP-Binding Protein, Ras-related (Rho) |
Bud1 (RSR1), RHO1, RHO2, RCN1 |
|
GTPase Activating Protein (Rab6-specific) |
GYP6 |
|
GTPase Activating Protein NF1 (Neurofibromatosis 1) |
IRA1, IRA2 |
|
Phosphatidylinositol 3-Kinase |
DRR1, TOR3, VPS34 |
|
PI-specific Phospholipase C |
PLC1 |
|
Ras-associated GTPase Activating Protein |
CLA2, (BUD2, ERC25) |
|
Vav (hematopoietic-specific GDP-GTP exchange factor) |
CDC24 |
The experimental procedure involves (i) targeted mutagenesis of the endogenous GPCR and Gα genes, (ii) covalent linkage of the human GPCR to yeast Gα by gene fusion to overcome the lack of recognition specificity between the human GPCR and the yeast Gα, and (iii) mutagenesis of the Gα domain and selection for functional coupling to ensure efficient interaction (Figure 1). The functional assays designed to enable convenient measurements and clear-cut read-outs are typically, but not exclusively, based on (i) growth arrest and morphological change to “shmoos”, (ii) auxotrophic selection, i.e. activation of the GPCR leads to expression of an essential gene which will support growth in minimal medium, (iii) color development, i.e. activation of the GPCR leads to expression of β-galactosidase, (iv) fluorescence, i.e. activation of the GPCR leads to expression of A. victoria green fluorescent protein (Figure 2).
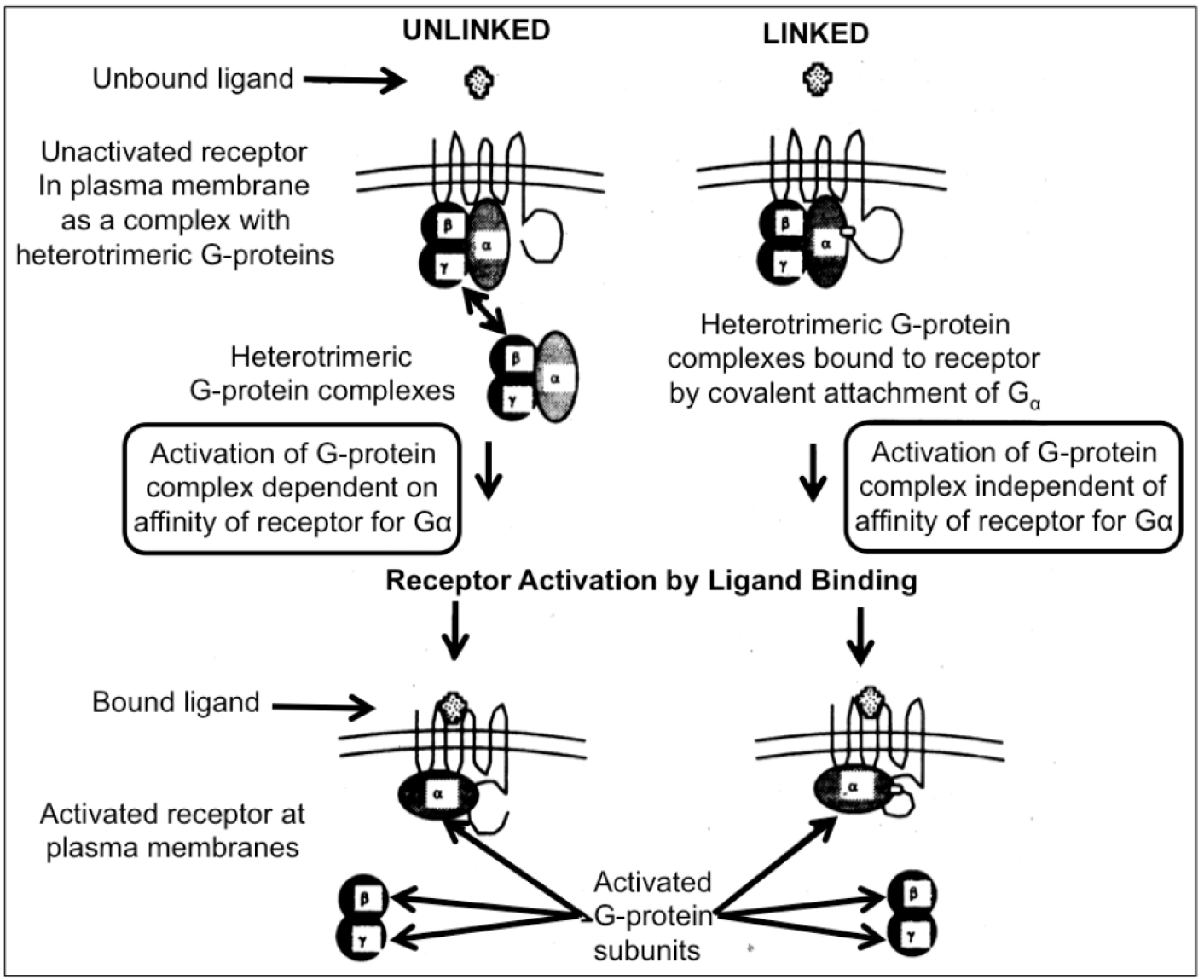
Figure 1. Two different modes of activation of heterotrimeric G-proteins by receptors are feasible in yeast cells. In the unlinked physiological mode of interaction between receptor and G-protein, the activation depends on the affinity between both partners (left section). A possible “mismatch” between a mammalian receptor expressed in yeast and the endogenous G-protein may be overruled by covalent linkage to Gα to the receptor. Nevertheless, the engineered covalent hybrid receptor-G-protein signaling complex in yeast is responsive towards activation/dissociation of the endogenous Gβ/Gγ-subunits by ligand binding to the mammalian receptor “subdomain” (right section).
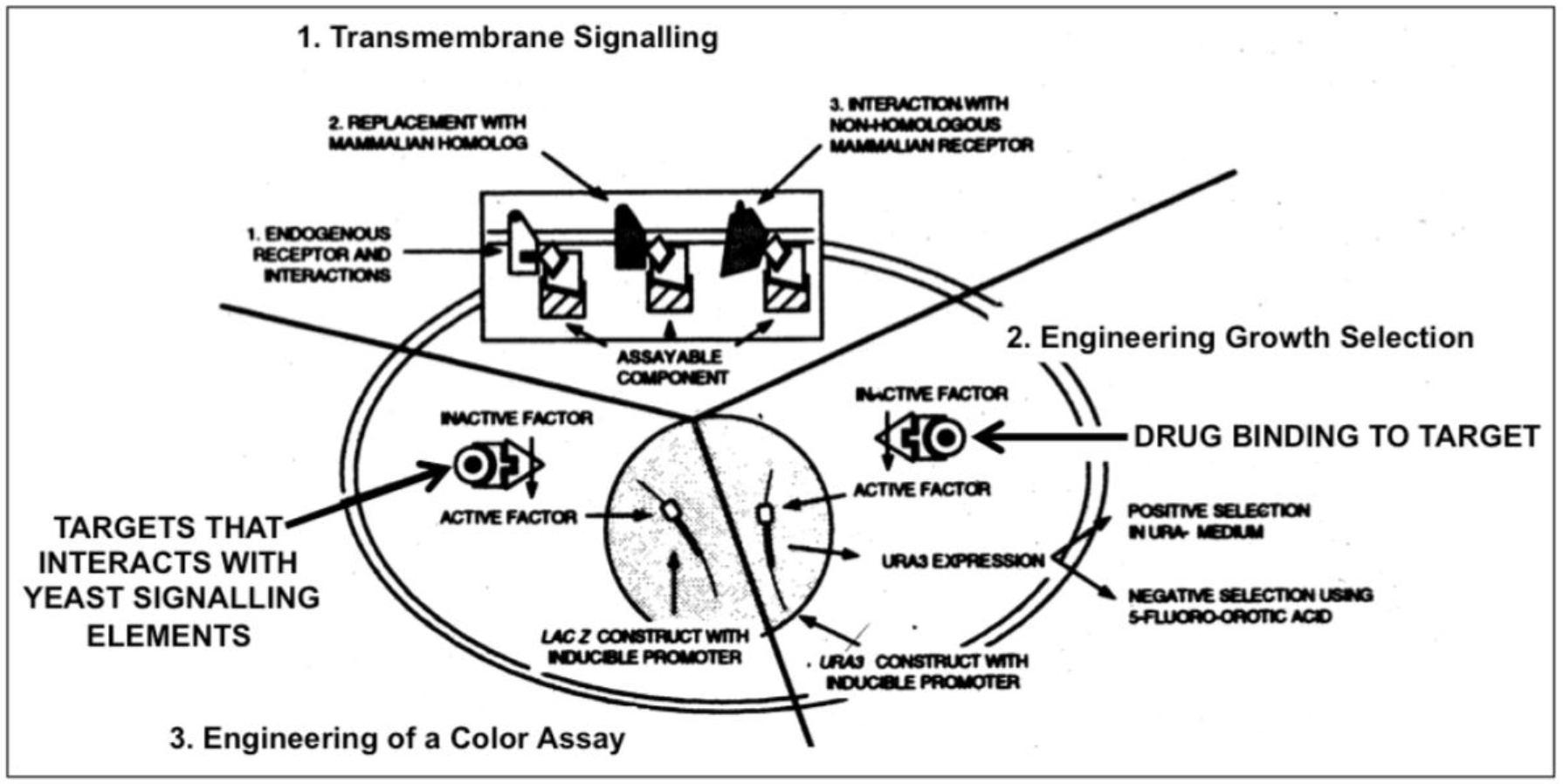
Figure 2. Engineering yeast for drug discovery requiries functional reconstitution of transmembrane signaling (1) which can be coupled to a signaling pathway regulating growth (2) or transcription of a reporter gene (e.g. ß-galactosidase) enabling a simple color assay (3).
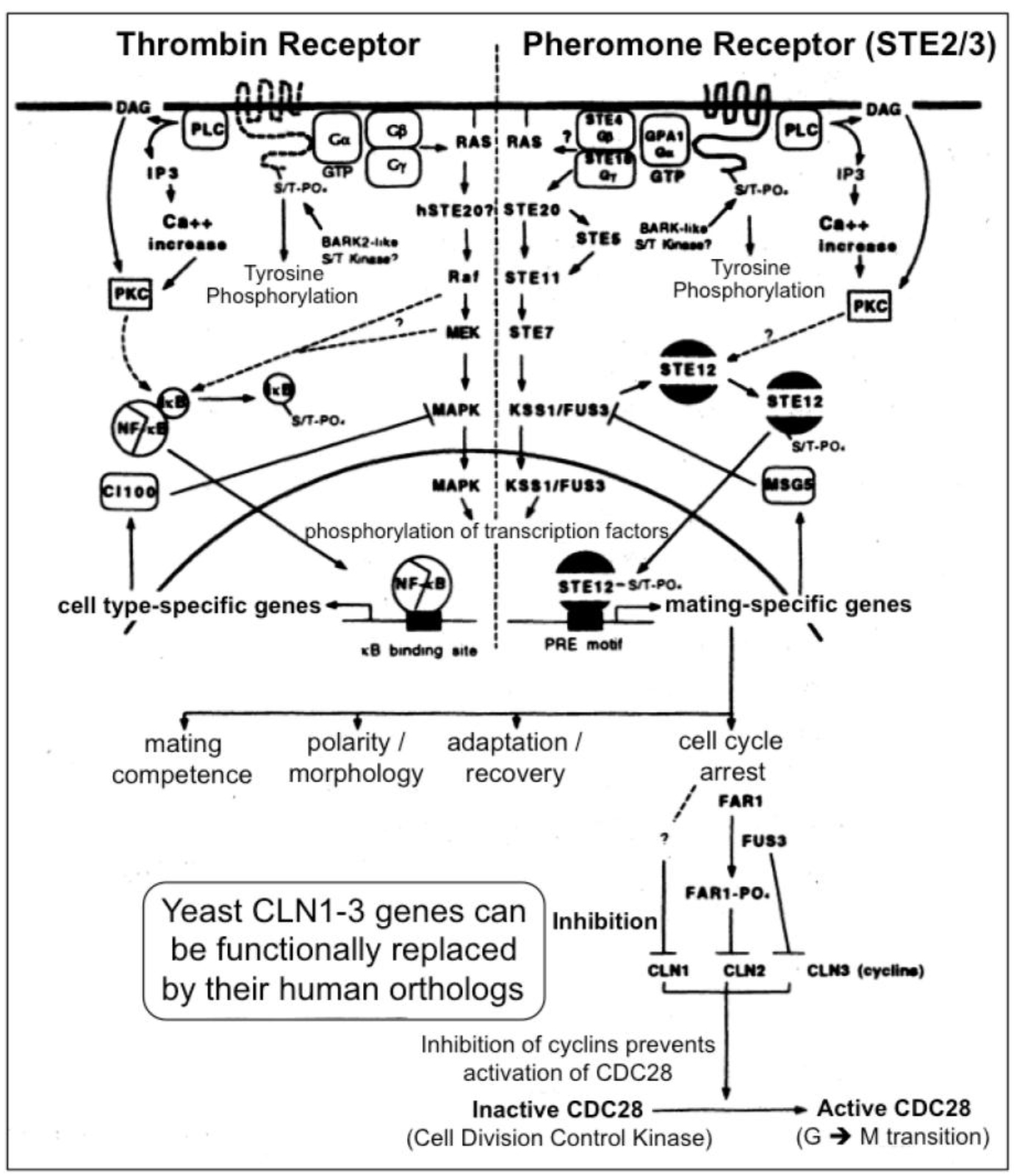
Figure 3. A multitude of orthologous components is engaged in signaling via heterotrimeric G-proteins in yeast (right section) and mammals (left section)
2.1. General Strategy
In yeast the heptahelical GPCR, Ste2, is engaged in intracellular signal transduction initiated by the α-factor pheromone (Figure 3) [12, 13]. Compatible with the evolutionarily conserved signaling function of Ste2, its expression in human HEK293 cells led to stimulation of the MAPK Erk1/2 upon incubation with α-factor [14]. Stimulation of Ste2 in yeast led to its endocytosis, ubiquitination and finally degradation, whereas in HEK293 cells, the α-factor-induced internalization of Ste2 was not accompanied by significant downregulation of the cellular amount of Ste2. The lack of expression of typical receptor tyrosine kinases, such as the insulin receptor and IGF-1 receptor, in yeast makes Ste2 a perfect target for the identification of the protein motifs which enable the regulation of GPCRs through mammalian tyrosine kinases and vice versa for the impact of GPCR expression and activation on receptor tyrosine kinase signaling, such as desensitization of the insulin receptor as the molecular mechanism for insulin resistance and T2D. For instance, in the basal state a Ste2-GFP fusion protein becomes targeted to the plasma membranes, but does not undergo endocytosis upon challenge of the yeast cells with insulin, as is typical for the GPCR ß2-adrenergic receptor in course of agonist binding [14]. The failure of insulin to control the intracellular trafficking of Ste2 enabled the construction of a model system consisting of yeast Ste2 and mammalian cells for studying the protein motifs which are responsible for the “linear” or “single-hit” GPCR receptor biology (i.e. agonist binding > receptor activation > endocytosis > ubiquitination > proteasomal degradation) induced by typical GPCR ligands, such as ß-adrenergic agonists. In higher eukaryotic cells, such as insulin target cells (liver, muscle, adipose), the resensitization and recycling of endocytosed GPCRs is typical, which raises the question about the molecular mechanisms controlling the recycling efficacy and thereby the periods of cell-surface retention. Yeast GPCRs are known to operate independent of cross-talk from/to receptor tyrosine kinases, which could influence the cell surface expression of GPCRs in mammalian cells. However, in yeast the endocytosis of GPCRs is not coupled to their rapid degradation or recycling back to the cell surface. The molecular mode of the control of GPCR trafficking by receptor tyrosine kinases, such as the insulin receptor, turned out to encompass the direct tyrosine phosphorylation of the GPCR as well as the serine/threonine phosphorylation by downstream protein kinases, such as PKB/Akt. Thus the identification of the complete panel of amino acid motifs, phosphorylation sites and downstream binding events with adaptor proteins, such as Grb2, can be achieved by the recombinant introduction of selected regions of the mammalian GPCR of interest into yeast Ste2 and subsequent assaying for the fate of this chimeric receptor in response to the corresponding agonist in animal cells. Thereby it was previously shown that the downregulation by degradation of a chimeric yeast Ste2 upon challenge with insulin is conferred by substitution of the endogenous cytoplasmic domain of Ste2 with that of the ß2-adrenergic receptor [14]. This model system will enable the screening for protein kinases, protein phosphatases and adaptor molecules which are engaged and required in the recycling/resensitization of a mammalian GPCR, such as ß2-adrenergic receptor, under the control of a mammalian tyrosine kinase, such as the insulin receptor, and vice versa, of a tyrosine kinase under the control of a GPCR. Thus, the investigation of such endocytosed Ste2 chimera may be useful for the elucidation of novel drug targets as well as insulin-mimetic and insulin-sensitizing agents for the therapy of diabetes.
2.2. GLP-1 Receptor as Target
The finding that a more pronounced insulin secretion results from the administration of glucose via the oral compared to the intraveneous route prompted the postulation of the action of so-called incretins [15]. GLP-1 is a members of the incretins which is secreted by intestinal cells in response to nutrient ingestion. GLP-1 contributes to the regulation of blood glucose predominantly via the induction of insulin release from pancreatic ß-cells [16]. GLP-1 triggers this insulin-releasing effect by binding to and activation of a typical GPCR, the GLP-1 receptor [17]. Thereby, the GLP-1 receptor plays an important role in glucose-dependent insulin release and has gained major interest as a target for the identification of novel (insulin-releasing) drugs for the therapy of T2D [18]. Interestingly, T2D is not only characterized by reduced insulin sensitivity and capacity of insulin secretion, but by decreased serum concentrations of GLP-1, in addition [19]. Strikingly, the intravenous injection of GLP-1 peptide causes potent upregulation of glucose-dependent insulin secretion and counteracts hyperglycemia in T2D patients [20]. Unfortunately, the plasma half-life of GLP-1 (7–36 amide) as the active version of GLP-1 is very short due to rapid cleavage to the inactive version GLP-1 (9–36 amide) by the serine protease dipeptidyl peptidase IV [21]. To overcome this limitation and to improve plasma half-life, a number of DPP-IV-resistant peptidic GLP-1 analogues, such as exenatide and liraglutide (Table 4), were developed which meanwhile have been introduced into the therapy of T2D [22].
In fact, these GLP-1 mimetics turned out to have important advantages in the control of T2D, such as rapid weight loss and low risk for hypoglycemic episodes, but their chronic application has been implicated with elevated risk of cancer and pancreatitis [23]. Unexpectedly, different GLP-1 mimetics sharing pronounced sequence homology exhibit considerable differences in their clinical profile [24]. This heterogeneity may be explained by the engagement of multiple isoforms of the GLP-1 receptor or, alternatively, the differential activation of the GLP-1 receptor with regard to its ability to coupling to and activate distinct G-proteins and thereby to induce distinct downsteam signaling pathways. Thus, much remains to be learnt about the biology of the GLP-1 receptor, in particular the (different) mode(s) of interaction of ligands and the resulting differential consequence(s) for the downstream responses. This inadequate and incomplete understanding of the molecular mechanisms of GLP-1 action in combination with the concerns regarding the safety of GLP-1 mimetics led to their categorization as “tier 2” of the consensus algorithm for initiation and adjustment therapy for the American Diabetes Association and European Association for the Study of Diabetes. Under this category the “less well-validated” treatments are encompassed and recommened as the last-line treatment of patients. This judgement can only be revised on the basis of a better knowledge of the differential activation and downstream signaling of the GLP-1 receptor in course of ligand binding. Corrresponding studies should facilitate the development of GLP-1 mimetic drugs with improved efficacy and safety.
The conventional assays used so far for studying GLP-1 receptor signaling are affected by other signaling pathways which manage to cross-talk to the receptor. Recently a robust and simple yeast-based assay system has been introduced which enables the monitoring of single GPCR-G-protein couplings with the aim to decipher the impact of a defined G-protein subunit on GLP-1 receptor downstream signaling [26]. Previously established test systems for the measurement of the dissociation constants of agonists and antagonists as well as relative binding efficacy of agonists have demonstrated the robustness of signaling assays using Saccharomyces cerevisiae cells [27]. Even more importantly, since the GPCR-G-protein interaction is defined in unequivocal fashion, the putative bias of a given (expressed) GPCR or G-protein for the interaction with a certain G-protein or GPCR, respectively, and corresponding consequences for downstream signaling can be elucidated rather conveniently without confounding effects potentially caused by other GPCRs and competing G-proteins. Subsequently, this yeast signaling assay has been adapted to establish the G-protein bias profiles for a number of GLP-1 mimetics (Table 4) [28].
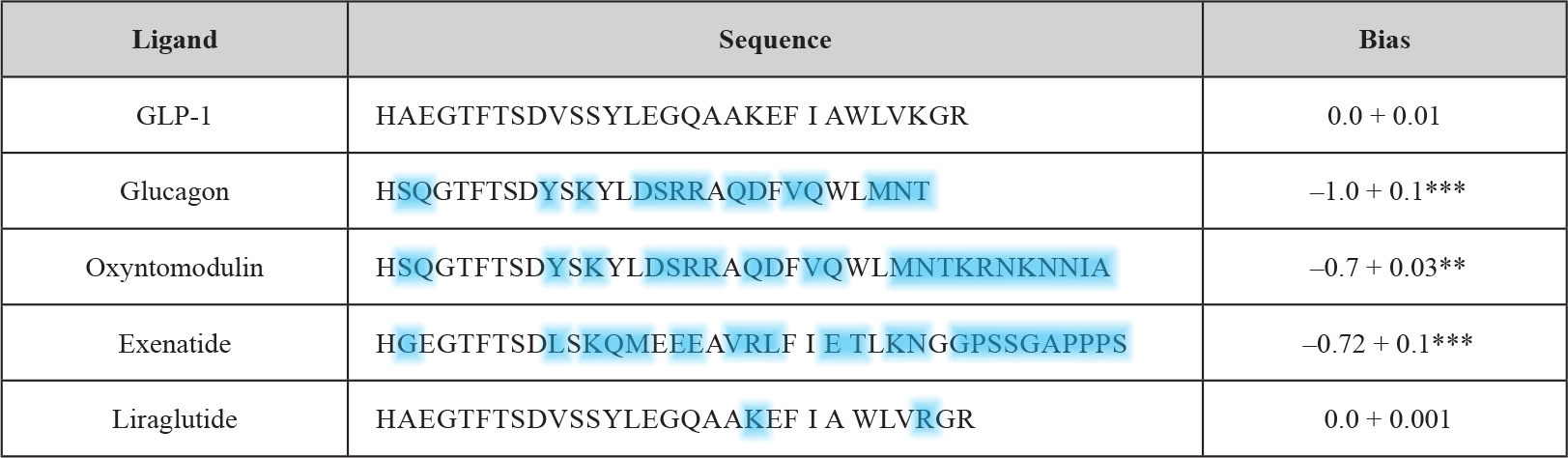
Table 4. Comparison of GLP-1 receptor peptide ligands and relative bias factors (adapted from and modified according to Weston 2014). The sequences of the various peptide ligands for the GLP-1 receptor are aligned to the natural agonist (GLP-1). Deviations in amino acid sequence from GLP-1 of the other GPCR ligands are highlighted in blue. The relative (to GLP-1) bias factor was quantitatively evaluated for each ligand as the change in log(τ/KA) ratio where a negative value indicates preference for the inhibitory Gαi chimera. Statistical significance was determined using one-way ANOVA with Bonferroni’s post-test with each data set compared with GLP-1 (**P < 0.01, ***P < 0.001). Data are mean of 5–8 independent experiments + SEM.
The use of yeast compared to other (e.g. mammalian) test systems has the benefit of providing a relatively zero background for G-protein activation which allows the identification of activation profiles at the individual level. This is of particular importance for the GLP-1 receptor since it is known that this receptor like other GPCRs is known to couple to a multitude of different G-proteins. The predominant one is the Gαs subunit which upon activation signals for the stimulation of cAMP generation. A minor one is the pertussis toxin-sensitive inhibitory Gαi [29, 30], the functional outcome of this coupling however remains to be characterized in greater detail [31]. In fact, the productive coupling of GLP-1 to Gαi was confirmed [28] with the use of the yeast chimeric Gα system [26, 32, 33]. GLP-1 as the natural peptide ligand elicited the concentration-dependent induction of the GPA1/Gα1 chimera with significantly diminished potency (EC50) and efficacy (log τ) in comparison to signaling through GPA1/Gαs. Interestingly, antagonism of the signaling event was left unaltered. These findings can be interpreted with the G-protein subunit expressed being irrelevant for the antagonist affinity and the measured alterations in agonist affinity being due to the preference for a certain G-protein, but independent of the use of the yeast assay system. Significant differences between the two systems were not measured with regard to the dissociation constant for the receptor antagonist, exendin-3. In contrast, for the therapeutically relevant GLP-1 ligands, liraglutide and exenatide, considerable but previously unrecognized differences in G-protein signaling were detected with exenatide exhibiting a pronounced bias for the Gαi pathway. Subsequently, this approach was extended to the investigation of small-molecule allosteric compounds and the closely related GPCR glucagon receptor. In conclusion, the yeast GPCR assay system allows the reliable and system-independent measurement of the pharmacological properties of putative GPCR-based drugs. The glucagon receptor as well as the GLP-1 receptor belong to the secretin (family B, class 2) GPCR family with 15 members. The peptidic ligands for those receptors typically consist of 27 to 84 amino acids.
2.3. The Use of Permeable Yeast Cells
For decades the investigation of GPCRs in yeast was not favored by the researchers since the yeast cell wall was thought to prevent their naturally occurring peptidic ligands from reaching the (periplasmic face of the outer) plasma membranes, where the ligand binding domain of the (ectopically) expressed GPCRs will be located. However, recently the ectopic expression of two corticotropin-releasing factor receptor subtypes [34, 35] as well as the functional co-expression of the calcitonin receptor-like receptor together with a variety of distinct receptor activity-modifying proteins (RAMPs) [36] has been demonstrated. In apparent conflict with the above assumption of the non-permeation of GPCR ligands across the yeast cell wall, in those cases as is true for the glucagon receptor and the GLP-1 receptor, the yeast-based assay system succeeded in the accurate reproduction of the in vitro pharmacology (e.g. relative binding affinities, G-protein coupling), albeit with absolute binding affinities typically below those measured in corresponding mammalian test systems. Nevertheless or to overcome the latter limitation permeable yeast cells have been developed.
Microbial cells have evolved an impermeable cell wall and plasma membranes that allow them to survive in the environment. Therefore the drug target may not easily accessible to compounds in high-throughput screens that use microbial cells, which in consequence are likely to miss those compounds being unable to penetrate across the cell wall and plasma membrane barriers. In addition, many microbes have very effective efflux systems that pump out compounds. These efflux systems are similar to the multidrug resistance (MDR) transporters found in tumor cells. One of the large classes of efflux systems, or transporter, is called the ATP-binding cassette transporters or ABC transporters. The ABC transporters are conserved fom bacteria to man [37]. With such drawbacks, can microbial- and yeast-based screening be effectively used for drug discovery? Genetic and molecular technology has made it possible to remove some of these barriers and make screen development and screening in unicellular lower eukaryotes a viable, inexpensive, and productive alternative to other screening systems. Among lower eukaryotes, Saccharomyces cerevisiae have been the most popular because of the genetic manipulations feasible with this organism. Since it is an eukaryote, it is often considered to be a more realistic system for screening for mammalian drug targets as compared to prokaryotes, such as E. coli. However, yeast is slower to grow than E. coli, taking 48 h to grow to adequate cell densities, while E. coli can be used within 6 to 8 h of growth. The genetic manipulations in E. coli are considerably less difficult than with yeast and E. coli are more permeable than yeast.
In fact, wild-type Saccharomyces cerevisiae is quite impermeable owing to their cell wall and plasma membranes. The cell wall is considered to be latticelike and allows most small molecules to permeate through. However, the plasma membranes are considered to be quite impermeable. Strikingly, previously it has been noted that yeast cells are actually permeable and the lack of drug effect is the result of the activity of multiple efflux systems, belonging to the family of ATP-binding cassette transporters (MDR), called PDR, that rapidly pump out compounds. The transcription factors, pdr1p and pdr3p, down-regulate the expression of hexose transporters, HXT11 and HXT9, which in turn up-regulate the expression of PDR (Figure 4). Thus deleting the hexose transporters, HXT11 or HXT9, confers pleiotropic drug resistance on yeast while overexpression of these transporters results in increased sensitivity to drugs. Furthermore, deletion of the regulators of the promoter for the ATP-binding transporters, PDR1 and PDR3, in HXT11 and HXT9 over-expressing strains, results in supersensitive yeast [38]. These mutant strains are ideal organisms for use as host strains for the development of screens. Improved cell permeability was also reported for the yeast strains with deletion of the YOR1 gene, which encodes another ATP-binding cassette transport protein [39]. In addition to transporter mutants, mutants in the ergosterol pathway, such as in ERG6, are also more permeable to small molecules used in screening compared to wild-type strains [40]. However, the mutants in the ergosterol pathway have the disadvantage that their growth is negatively affected, and they are difficult to transform.
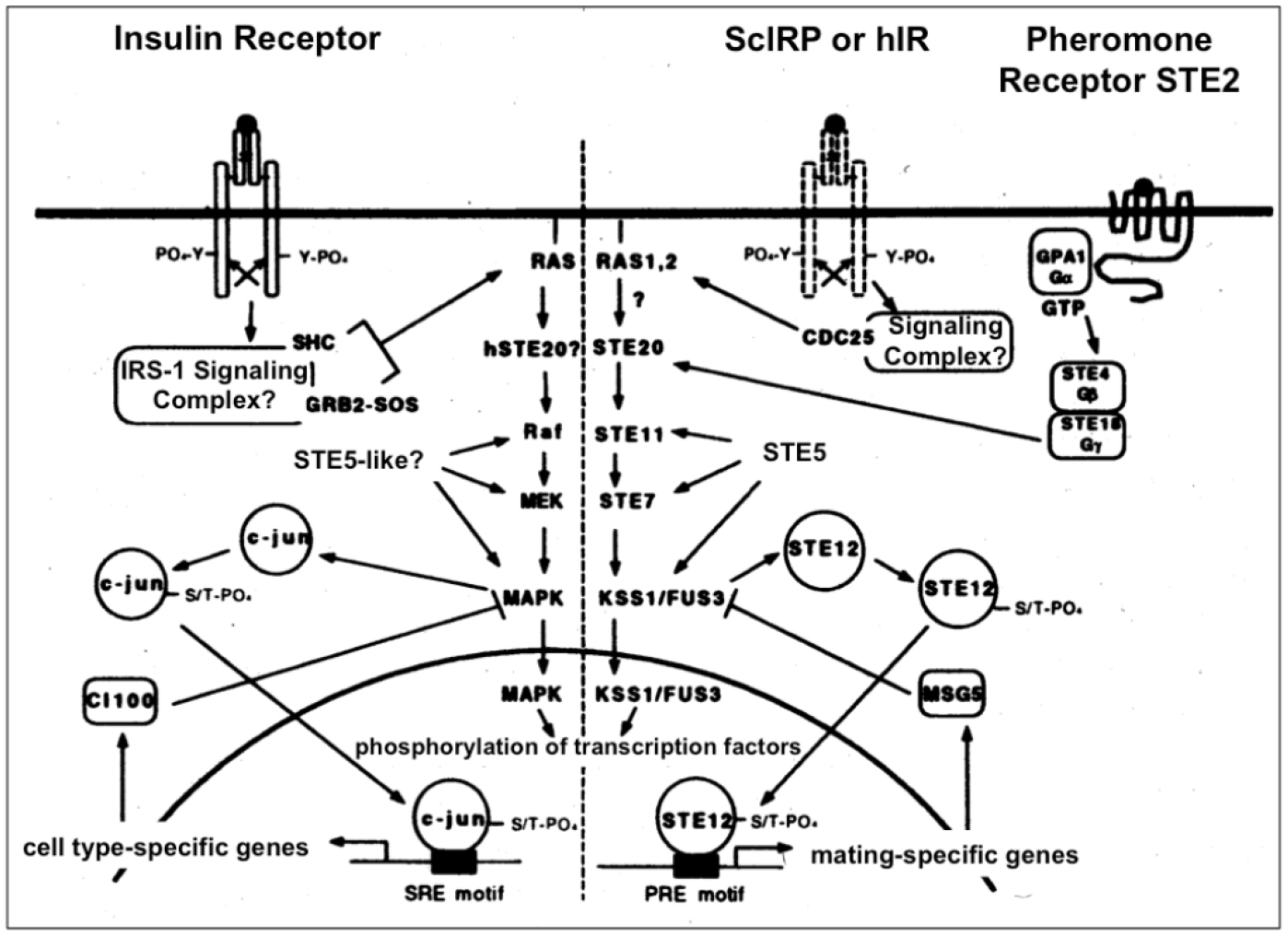
Figure 4. Insulin signal transduction may regulate the MAPK cascade in yeast (right section). Insulin signaling in insulin-responsive mammalian cells via the homologous Ras-MAPK pathway is shown for comparison (left section). In yeast coupling of the endogenous (ScIRP) or heterologously expressed human (hIR) insulin receptor to the MAPK pathway could occur via the heterotrimeric G-protein, GPA1/STE4/STE18 to STE20 (which is also used by the pheromone receptor during the mating response) or via CDC25 to Ras1/2, respectively, which may be analogous to mammalian cells where the insulin receptor transduces its signal to Ras via the GDP-GTP exchange factor SOS.
It would be useful and feasible to extend this technology for the inclusion of many GPCRs or other components, which putatively interact with the receptor in situ (e.g. regulatory proteins of G-protein signaling, RAMPs), in order to recognize any (positive or negative) effect of an interaction built up on the receptor pharmacology and the resulting (patho)physiological changes. Moreover, the relative simplicity and convenience of the ectopic expression of GPCRs in yeast would be compatible with the introduction of patient-specific mutations into GPCRs with the aim to study and to quantitatively analyze their impact on the ligand-induced signaling capability of the receptor and possibly to efficiently screen for ligand mimetics, which bypass the defect. Thereby, the yeast-based assay system could contribute to the development of personalized drugs.
3. Insulin Receptor and Signaling as Target
This approach originated during an attempt to study the molecular basis for tyrosine kinase-linked signal transduction in a cell system in which there is no detectable endogenous receptor tyrosine kinase-coupled regulatory processes, namely, the fission yeast Schizosaccharomyces pombe. Expression of selected mammalian signaling elements in such a naive system might support productive interactions between heterologous components while eliminating involvement and cross-talk with host cell tyrosine kinase-associated control systems. S. pombe has been used by others to study the function and interactions of selected mammalian signaling elements, including PKC isoforms, Src, Csk, Raf-1, Ras, PI-3’K and MAPKK. Furthermore, high level expression of the platelet-derived growth factor receptor and its substrate phospholipase Cγ (PLCγ) in S. pombe led to autophosphoryation and substrate phosphorylation at tyrosine residues. Therefore, appropriate cleavage and oligomerization of the expressed human receptor seem to be possible. Potential assays may rely on (i) modulation of growth, (ii) insulin binding, (iii) autophosphorylation of the receptor at tyrosine residues, (iv) tyrosine phosphorylation of heterogenously expressed authentic or engineered substrates, such as IRS-1 and PI3K as well as fusion proteins or fragments derived thereof, or artificial substrates, e.g. EEEY and (v) phosphorylation of yeast substrates (Figure 4). These experimental designs should demonstrate, that heterologous expression of selected signaling elements in yeast will permit the detection and analysis of functional insulin receptor activation and pave the way for further systematic and detailed molecular dissection of a wide range of interactions, central to insulin receptor kinase-coupled signal transduction processes as well as for the identification of peptidic or small peptidomimetic molecules, which manage to strengthen or disrupt those interactions.
Alternatively, the assays for human receptor activation may be based on signal transduction pathways activated by receptor tyrosine kinase activity: I. The soluble domain of the human insulin receptor can be stably expressed at the plasma membranes of Saccharomyces cerevisiae [42]. II.Tyrosine phosphorylation of some yeast proteins is increased upon incubation of intact cells or spheroblasts with human insulin [43]. This raises the possibility that the tyrosine kinase activity of the human insulin receptor may initiate signal transduction cascades leading to phosphorylation of endogenous substrates, and their human orthologs expressed in yeast. This will require (i) cloning of the yeast signaling proteins that respond to the human insulin and insulin-dependent (tyrosine/serine/threonine) phosphorylation, (ii) identification and (iii) expression of their human orthologs. III. Low levels of expression of the insulin receptor kinase domain may lead to a very modest but reproducible slowing of growth, high expression may result in stronger inhibition as has been reported previously for overexpression of the constitutively active human insulin receptor in cultured CHO cells [44]. In general, clear-cut effects of activated yeast or human receptors on downstream signaling proteins of yeast or human origin represent potential parameters of measurement for a yeast-based screening assay. Taken together, yeast and other lower eukaryotic unicellular organisms have already proven their usefulness in various aspects of pharmaceutical development and, no doubt, their impact will further increase during the next years.
4. Endoplasmic Reticulum Stress and Pancreatic ß-Cell Dysfunction as Target
Failure of insulin secretion caused by reduction in the number and functionality of pancreatic ß-cells has been considered as the hallmark of T2D for decades [45, 46]. The majority of T2D patients is characterized by overweight or obesity which in general is correlated to elevated levels of plasma free fatty acid (FFA) levels [47, 48]. Albeit being still a matter of intense debate, this hyperlipidemic state could be (causally) involved in the loss of functional ß-cells through a pathophysiological process called lipotoxicity [49]. Importantly, the operation of lipotoxic processes under conditions of overfeeding and excessive calorie uptake, as often prevalent during obesity and T2D, has also been reported for a number of non-ß-cells, such as adipocytes, myocytes, hepatocytes and brain cells, and linked to their desensitization towards insulin challenge [50–53]. The molecular mechanisms underlying the FFA-induced lipotoxicity have been investigated in vitro during the past two decades in course of direct incubation of cultured ß-cells, pancreatic islets or insulin target cells and, more recently, of yeast cells (Saccharomyces cerevisiae) [54–56]. Strikingly, despite the tremendous differences between these cell types considering their morphology, structure and physiology, they apparently display pronounced similarity with regard to their susceptibility and viability towards FFA (Table 5). This argues for conservation, at least in part, of lipotoxic mechanisms along the evolution of eukaryotes from yeast to humans. Importantly, in ß-cells, insulin target cells and yeast cells the extent of the lipotoxic effects provoked by the FFA critically depends on the number of their carbon atoms and the degree of their saturation. The data available so far are consistent with saturated (SFA) and long-chain FFA (e.g. palmitate C16:0, stearate C18:0) exerting the most adverse effects, whereas saturated and short(er)-chain ones (e.g. myristate C14:0 and below) as well as long-chain unsaturated fatty acids (UFA) equipped with one to several double bonds (e.g. palmitoleate C16:1, oleate C18:1, linoleate C18:2 and linolenate C18:3) exhibiting considerably lower harmfulness [57–59]. In addition, the concentration-dependent abrogation of the SFA-triggered lipotoxic effects, in general, and of cell death, in particular, by excess of UFA has meanwhile been amply documented [60, 61].
Most importantly, in both ß-cells and yeast cells the SFA-triggered lipotoxicity leads to the initiation of the so-called stress response in the endoplasmic reticulum (ER) or unfolded protein response (UPR)(Table 5). The ER represents the site for a number of essential physiological processes, such as Ca2+-homeostasis, (phospho)lipid biosynthesis and biogenesis of proteins, which have to be transported to intracellular organelles and the plasma membranes or secreted into the extracellular milieu. Prior to guidance into the secretory pathway the membrane and secretory protein precursors have to be correctly folded and assembled in the ER. Any imbalance between the folding capacity of the ER and its passenger protein load will ultimately cause ER stress [62]. ER stress has to be counterbalanced by the multiple strategies of the UPR, such as proteolytic degradation [63] to preserve or re-gain the structural and functional integrity of the ER despite the accumulation of misfolded polypeptides [64, 65]. It is believed that the SFA-triggered cell death is mediated through ER stress and the failure to cope with it in the presence of high concentrations of SFA, which finally will result in apoptosis.
Table 5. The effects of exposure of yeast- and ß-cells towards palmitate in comparison.
|
UPR pathway |
Apoptosis/ Growth arrest |
Misfolded proteins |
Depleted ER Ca2+ stores |
Altered ER morphology |
|||
|
Ire1 |
PERK |
ATF6 |
|||||
|
ß-cells |
+ |
+ |
+ (?) |
+ |
+ |
+ |
+ |
|
yeast |
+ |
None |
None |
+ |
+ |
not tested |
+ |
In higher eukaryotes, a critical pathway engaged by the UPR, which senses for unfolded membrane protein precursors, is constituted by the inositol-requiring enzyme 1 (IRE1) cascade [65], which in ß-cells seems to be upregulated in response to long-chain SFA, such as palmitate. The IRE1 cascade constitutes the only UPR pathway which is conserved from yeast to humans. In Saccharomyces cerevisiae the accumulation of misfolded proteins triggers dimer formation of Ire1p which results in upregulation of its endogenous endoribonuclease activity. In non-stressed yeast and ß-cells the ER-resident protein of the HSP70 family, Kar1p and BiP, respectively, binds to Ire1(p) for suppression of its activation. In stressed cells harboring excess of misfolded proteins in the ER Kar1p / BiP manages to dissociate from Ire1(p) and thereby enables Ire1(p) dimerization which finally leads to its autophosphorylation and activation [64]. The Ire1p endoribonuclease activity is directed against the mRNA of the transcription factor Hac1p which interacts with the promoter of UPR elements (UPREs) and controls the expression of about 5% of the genes in yeast (and is the yeast ortholog of mammalian XBP1). Importantly, in yeast upregulation of the Ire1p cascade in the presence of high concentrations of SFA was observed using a reporter gene (LACZ) assay which monitored the transcription activity of four UPREs, i.e. the amounts of active Hac1p/XBP1, as reflected in the measured ß-galactosidase activity [66]. High intracellular concentrations of SFA in yeast as well as in ß-cells, which may result either from blockade of endogenous fatty acid desaturation or from the presence of palmitate in the incubation medium, have been reported to provoke full induction of the Ire1(p) cascade. Consistent with the beneficial effects of UFA, Ire1(p) activation was diminished in concentration-dependent fashion in the presence of oleate [67].
5. Adipocyte Lipolysis as Target
The concentration of FFA in the blood of mammals which are tightly linked to their metabolic disease state, such as T2D in case of chronically elevated levels, critically depends on the efficacy of lipolysis in the various adipose tissue depots [68]. Insulin with its primarily anabolic function in the body manages to restrict lipolysis and to foster the incorporation of FFA, which are ultimately derived from the ingested nutrient lipids, into neutral triacylglycerol, which becomes stored in adipocytes, during the postprandial state. Missing suppression of adipocyte lipolysis by insulin has been considered since decades as the major physiological defect which is causative for metabolic diseases, such as T2D and obesity, through the induction of insulin resistance in peripheral tissues and dysfunction and death of ß-cells through the operation of lipotoxic mechanisms (see above) [69–71].
Triacylglycerol is lipolytically degraded to FFA and glycerol through the joint action of tri-, di- and monoacylglycerol lipases [72–75] with the most recently unraveled Adipose Triglyceride Lipase (ATGL) [76–78] exerting the major portion of the triacylglycerol hydrolase activity in adipose, muscle and liver tissues and representing the rate-limiting enzyme for lipolysis. This is manifested in the observation that up- and downregulation of ATGL expression unequivocally leads to increase and decrease, respectively, of both the basal and the cAMP-induced triacylglycerol hydrolysis [79–81]. It has been accepted for decades that the rate of lipolysis is primarily under the short-term and post-translational control of the cAMP-dependent signaling pathway through phosphorylation of the lipid droplet coat protein perilipin and the diacylglycerol lipase HSL by PKA leading to rapid stimulation of lipolysis, whereas insulin-induced blockade of cAMP-dependent signaling via Akt-dependent [82] and -independent [83] mechanisms causes inhibition of lipolysis.
In addition, insulin and other physiological anti-lipolytic stimuli, such as feeding and α-adrenergic hormones, have to elicit a long-term inhibitory effect on lipolysis which is based on the suppression of its rate-limiting enzyme, ATGL, most likely as a consequence of downregulation of ATGL expression. Recently, a novel molecular mechanism for the negative regulation of ATGL expression by insulin and nutrients has been identified, the mTORC1-dependent pathway which blocks lipolysis by reduction of ATGL transcription [84]. Interestingly, in Drosophila a similar mechanistic link between dTORC1 and the ATGL homologue Brummer lipase [79, 85] was demonstrated recently [86, 87], which even can be extended to the insulin receptor [88]. The apparent evolutionary conservation of this anti-lipolytic molecular mechanism strongly argues for its essential physiological role in eukaryotes, in general, and in humans, in particular, as well as makes it to an attractive source for targets for the therapy of metabolic diseases.
For elucidation of the molecular mode of mTORC1 action, yeast as a model organism was used based on the knowledge, that Saccharomyces cerevisiae harbors a functional ortholog of ATGL, the triglyceride lipase Tgl4p (Table 1) [80]. The putative involvement of the Tor1-dependent pathway in the expression of Tgl4 was studied by growing S. cerevisiae in the presence of the specific Tor1/mTORC1 inhibitor, rapamycin [81]. In fact, rapamycin led to a considerable and specific upregulation of the Tgl4p mRNA expression. For identification of the transcription factors engaged in Tgl4 expression, a S. cerevisiae deletion library was screened [81]. Interestingly, yeast cells lacking Msn4 were found to have increased basal levels of Tgl4 mRNA and to be almost insensitive towards the stimulatory effect of rapamycin on Tgl4 expression. The residual and very moderate positive effect of rapamycin on Tgl4 transcription in Msn4-defective yeast cells argues for the participation of factors other than Msn4 in the transcriptional control of Tgl4 expression by TOR1. Strikingly, orthologs of yeast Msn4p in mammals were reported to constitute a family of early growth response transcription factors, among them the well-known members Egr1 (Krox24), which are known to play important roles in adipocyte differentiation [89, 90] and regulation of cholesterol biosynthetic gene expression [91–93], and Egrs (Krox20) [94]. These data strongly suggest that the regulation of ATGL transcription by the mTORC1-Egr1 pathway is important for the control of lipid metabolism. Otherwise it would not have been conserved along evolution from yeast via Drosophila [86, 87] to mammals [84].
6. Use of Yeast for Drug Target Identification and Validation
Although yeast-based systems are less complicated than mammalian cell-based systems, orthologs of many mammalian proteins are found in unicellular lower eukaryotes. In addition, many other mammalian proteins that do not have sequence homology but do have functional homology can be used to complement functions in yeast. Unlike in higher eukaryotes, the functions of about half of the yeast genes are known on the basis of amino acid sequence similarity with other proteins of known function [95, 96]. This is an enormous resource that is being used for functional analysis. History has shown us that biological mechanisms revealed from the study of lower eukaryotic cells will be applicable to higher eukaryotes. In the future, knowledge of the function of yeast proteins will help in elucidating the function of many mammalian proteins. The similarity between living organisms was noted by Jacques Monod when he said “What is true for Escherichia coli is true for the elephant, except more so” [97]. In the simplest approach, functional complementation can be used to derive a screen in which the activity of the heterologous gene is essential for survival. This approach has been successful even in cases where protein homology is limited, as long as the relevant biological activity is complementary. Yeast-based systems can also be used to define interactions with other proteins. The more difficult approach is to manipulate the heterologously expressed gene to obtain a surrogate phenotype and create “designer yeast”.
6.1. Complementation of Homologous Target Proteins
Many currently used drug targets for the therapy of T2D are GPCRs, such as the glucagon and GLP-1 receptors (see above). Antagonists have been identified by ligand- displacement assays using mammalian cells or their membrane preparations. Agonists have been generally been identified by functional assays. Yeast-based systems have been adapted to identify agonists and antagonists of GPCRs. The mating factor receptor in S. cerevisiae, Ste2, is similar in structure to mammalian GPCRs. Mammalian GPCR can be used to replace Ste2, so that the GPCR can signal through the mating factor signaling pathway when activated by the GPCR agonist [98]. In order to get efficient downstream coupling with the mating factor pathway kinases, the amino-terminal domain of the Gα protein was deleted [99]. The GPCRs expressed in this manner in yeast are useful for screening for agonists. Coupling can also be obtained with the natural yeast Gα protein [100, 101].
The natural ligands of GPCRs can be large peptide ligands, such as for the GLP-1 receptor (see above). In mammalian cells, these GPCRs can be activated by these peptides as well as their derivatives. Antagonists are identified by finding compounds that can displace these peptides. Short peptides can be coexpressed with GPCRs in yeast to develop functional antagonist screens as well as for identifying ligands for orphan GPCRs [102]. Orphan GPCRs are those receptors that have been cloned by sequence homology to known GPCRs but whose function and natural ligands are not known. Peptide libraries have been expressed with secreted sequence tags that are secreted across the plasma membranes of yeast where they come into contact with the GPCRs. Using these peptide libraries, those peptides that specifically interact with and activate the receptor in an autocrine fashion can be identified.
Potassium channels are also important drug targets for the therapy of T2D, among them the ATP-dependent potassium channel (K+-ATP) constituted by pore-forming Kir and ligand-binding SUR subunits [103, 104]. Functional screens for K+ channel openers and blockers involve expensive equipment and are technically difficult to perform [105]. Therefore simpler assays for developing high-throughput screens have been appreciated. A simple functional screen was developed in S. cerevisiae using complementation of the TRK1/2 potassium transporter knockouts [106]. The inwardly rectifying potassium channel IRK1 has also been expressed in yeast to complement the Trk transporter defect. In the strain expressing the IRK1 channel, the ion channel activity correlates well with the growth phenotype and with patch clamp experiments in Xenopus oocytes expressing these channels.
The influenza M2 channel has been expressed in S. cerevisiae [105]. The influenza M2 channel is a proton channel that is expressed in infected cells. Its function is to increase the acidity of the milieu in which the virus sheds its capsid. When expressed in S. cerevisiae, the M2 proton channel increases the permeability of yeast plasma membranes to ions resulting in loss of yeast cell viability. In order to develop a screen to find influenza M2 protein inhibitors, it was expressed from a galactose-inducible promoter. The screen was designed to find compounds that permit growth and rescue the cells from the permeabilizing effects of M2 protein when the growth medium is supplemented with galactose. Channel screens designed in S. cerevisiae have been useful for high-throughput screening. However, it has to be considered that yeast is slowly growing, and expression of channels in this microbe is difficult and time-consuming.
6.2. Expression of heterologous target proteins
Screens to find ligands for steroid hormone receptors such as retinoid receptors, which constitute an important class of drug targets for T2D and cardiovascular diseases, have been designed in S. cerevisiae [107]. Steroid hormone receptors occur intracellularly and are built up from a ligand-binding domain, a dimerization domain, and a transactivation domain. When ligands induce these receptors to homo- or heterodimerize, their transactivation domain binds the specific response elements and activates specific promoters. The dimerization of the steroid hormone receptor followed by binding and transactivation of specific promoters can be studied in yeast. Homodimerization has been demonstrated using retinoic acid receptors, thyroid hormone receptors, and estrogen receptors [108, 109]. Heterodimerization with the RXR retinoid receptor can also be demonstrated [107]. In this system, the RAR retinoid acid receptors respond to a number of retinoids, but RXR responds only to the RXR-specific 9-cis isomer of retinoic acid [107]. Because all mammalian cells have many representatives of the steroid hormone receptor family expressed naturally, yeast-based systems offer cells with a “null” background activity for studying specific interactions.
Tyrosine-specific protein kinases and phosphatases are important drug targets for the therapy of T2D. Cell-free assays have been popular for this class of targets since the enzymes are easily produced by recombinant means and tyrosine-specific phosphorylation of the natural or an appropriate artifical substrate protein is simple to detect using labeled ATP. An alternative method using Schizosaccharomyces pombe has been published that will find inhibitors that are non-toxic to yeast as well as cell-permeable [110]. The prototypic tyrosine kinase, Src, was expressed under the control of the inducible promoter. Induction of Src results in cell death, and growth rescue can be used for the identification of inhibitors. To adapt the screen for identifying phosphatase inhibitors, this system was modified by co-expressing tyrosine phosphatase on a second plasmid. When the kinase and phosphatase are coexpressed, the cell survives the detrimental effects of kinase expression. Tyrosine phosphatase inhibitors can be identified in this system by looking for compounds that selectively kill the strain co-expressing the kinase and phosphatase [110].
6.3. Detection and Analysis of Protein-Protein Interaction
Proteins carry out their function in most cases by interacting with other proteins. The yeast two-hybrid system, developed by Fields and Song [111], has revolutionized the study of protein-protein interactions. In this system, the transcription factor, GAL4 from S. cerevisiae, is used to set up the assay. GAL4 has two domains, a site-specific DNA-binding domain and an acidic region that is required for transcriptional activation. The DNA-binding and activation domains can be coded by separate genes as long as they are brought together in a heterodimer to reconstitute a functional transcription factor. The system is designed so that when GAL4 binds the GAL4-binding domain on the promoter, LEU2 and/or HIS3 are expressed. Functionally competent chimeric proteins can be made that consist of the DNA-binding domain fused to one protein of an interacting pair and the activation domain fused to the second protein of the interacting pair. Interaction of the proteins that are constructed as chimeras of the activating and DNA-binding domains allow the yeast to grow in the absence of histidine and leucine, thus providing a selective advantage. The yeast two-hybrid system is widely used for identifying homo- and heterodimerizing proteins as well as to develop screens to find compounds that can block two proteins from interacting with each other [112].
The yeast two-hybrid system has been modified to measure the dissociation of interacting proteins by using the URA3 reporter [113]. Yeast cells expressing URA3 can grow in medium without uracil. When 5-fluoroorotic acid (FOA) is introduced into the medium, URA3 expressing cells take up FOA and transform it into a toxic compound. Thus the expression of the reporter gene is toxic and provides a powerful selection procedure. This FOA system is used in the “reverse two-hybrid” system, providing a selective growth advantage and a more powerful system for screening. In the “reverse two-hybrid system”, the interacting protein is expressed inducibly, and only when the interacting proteins are blocked do the cells survive. GAL4 and LexA transcription factors are most often used in the yeast two-hybrid system. In two-hybrid screens, it is useful to have two separate reporter constructs to help in sorting “hits”. Reporters such as Leu2 and LacZ can be expressed in the same cell.
The yeast two-hybrid system has been used to develop screens for ligand-receptor interactions, including peptide hormone receptors, such as the GLP-1 receptor, and the receptor tyrosine kinases, such as the insulin receptor [114–116]. Specific and reversible ligand-receptor interactions between growth hormone and growth hormone receptor, VEGF and KDR, can be studied using the yeast two-hybrid system. Ligand-dependent receptor dimerization can also be studied using three expression plasmids in which the receptor is expressed as a fusion protein with both the DNA-binding protein as well as the activation domain. The ligand is expressed from a third plasmid. When the ligand binds the two receptors, the DNA-binding domain and activating domains are pulled together and GAL4 is activated.
The yeast two-hbrid system has been adapted to study protein-protein, protein-RNA, protein-DNA, and protein-small molecule interactions [117]. A one-hybrid system has been developed that utilizes cis-acting sequences to identify DNA-binding proteins that can initiate transcription [118]. A yeast three-hybrid was developed to study RNA-protein interactions that are especially useful for developing screens against viruses [119]. In this system, the hybrid RNA containing sites recognized by the RNA-interacting proteins links the two-hybrid proteins containing the DNA-binding and activation domains, respectively. The yeast two-hybrid system has been recently applied to find inhibitors of the N type calcium channel [120, 121]. Alternative screening techniques use mammalian cells to measure calcium channel activity with electrophysiological and spectrophotometric methods to measure calcium influx. These methods are labor intensive, difficult, and not compatible with high-throughput screening. In the yeast two-hybrid system, the interacting, regulatory portion of the α1 subunit of the channel fused to the Gal4 activation domain and the full length β3 subunit fused to the yeast Gal4 DNA-binding domain were expressed. The system could be adapted to find inhibitors of specific calcium channels by selecting the specific interacting domains.
7. Conclusions
Unicellular lower eukaryotes provide an alternate platform for high-throughput screening. Those systems are inexpensive to run and screens can be developed rapidly. Many modular systems are available that are adaptable to important classes of drug targets, such as GPCRs, single-transmembrane receptors such as growth factor tyrosine kinases and phosphatases and ion channels, which are of critical importance for the therapy of metabolic diseases, such as T2D. Functional screens are becoming necessary for developing screens for proteins whose biological functions are not yet known as well as on proteins that interact with new proteins. Because of the ease with which new targets can be explored, functional cell-based screens are becoming the preferred method for finding leads for drug discovery. Test systems based on unicellular lower eukaryotes provide simple and cost-effective means for the identification and validation of drug targets and, most importantly, for the discovery of novel drugs by functional screening.
As thousands of new potential drug targets from genomic information and protein interaction studies have meanwhile been identified, the future of screening is in using chip technology [122, 123]. Systems based on unicellular lower eukaryotes are especially suited for the delivery to microchips. Yeast is robust and is easy to handle and distribute. Thousands of yeast cells can be deposited on chips in a high-density formate. In addition, the surface charge on S. cerevisiae could be used to array the organisms into the desired formate. Currently, the limitation is the sensitivity of reading colorimetric reporters, and alternatives are being investigated. Biosensors and transducers could be used to detect thermal, immunologic, or optical changes [124, 125]. Glucose sensing amperometric systems are being used in clinical microbiology and could be developed for high-throughput screening [126]. Each of the microbial systems described can be adapted to use the reporter that is suitable for the high-density formate on chips for screening. Consequently, a yeast genetic system has been designed for the identification of small-molecule inhibitors of protein-protein interaction on the basis of nanodroplets [127].
Conflict of Interest: The author declares no conflict of interest.
References
- Morris AJ, Malbon CC (1999) Physiological regulation of G protein-linked signaling. Physiol Rev 79: 1373–1430. [crosref]
- Saad MJ, Velloso LA, Carvalho CR (1995) Angiotensin II induces tyrosine phosphorylation of insulin receptor substrate 1 and its association with phosphatidylinositol 3-kinase in rat heart. Biochem J 310: 741–744.
- Baltensperger K, Karoor V, Paul H, Ruoho A, Czech MP, et al. (1996) The beta-adrenergic receptor is a substrate for the insulin receptor tyrosine kinase. J Biol Chem 271: 1061–1064. [crosref]
- Kowalski-Chauvel A, Pradayrol L, Vaysse N, Seva C (1996) Gastrin stimulates tyrosine phosphorylation of insulin receptor substrate 1 and its association with Grb2 and the phosphatidylinositol 3-kinase. J. Biol. Chem 271: 26356–26361.
- Velloso LA, Folli F, Sun XJ, White MF, Saad MJ, et al. (1996) Cross-talk between the insulin and angiotensin signaling systems. Proc Natl Acad Sci U S A 93: 12490–12495. [crosref]
- Karoor V, Shih M. Tholanikunnel B, Malbon CC (1996) Regulating expression and function of G-protein-linked receptors. Progr Neurobiol 48: 555–568.
- Karoor V, Malbon CC (1998) G-protein-linked receptors as substrates for tyrosine kinases: cross-talk in signaling. Adv Pharmacol 42: 425–428. [crosref]
- Karoor V, Malbon CC (1996) Insulin-like growth factor receptor-1 stimulates phosphorylation of the beta2-adrenergic receptor in vivo on sites distinct from those phosphorylated in response to insulin. J. Biol. Chem 271: 29347–29352.
- Whiteway M, Clark KL, Leberer E, Dignard D, Thomas DY (1994) Genetic identification of residues involved in association of alpha and beta G-protein subunits. Mol Cell Biol 14: 3223–3229. [crosref]
- Leberer E, Thomas DY, Whiteway M (1997) Genetic identification of residues involved in association of alpha and beta G-protein subunits. Curr. Opin. Genet. Develop 7: 59–66.
- Leeuw T, Wu C, Schrag JD, Whiteway M, Thomas DY, Leberer E (1998) Interaction of a G-protein beta-subunit with a conserved sequence in Ste20/PAK family protein kinases. Nature 391: 191–195.
- Jenness DD, Burkholder AC, Hartwell LH (1983) Binding of alpha-factor pheromone to yeast a cells: chemical and genetic evidence for an alpha-factor receptor. Cell 35: 521–529.
- Blumer KJ, Reneke JE, Thorner J (1988) The STE2 gene product is the ligand-binding component of the alpha-factor receptor of Saccharomyces cerevisiae. J. Biol. Chem. 263: 10836–10842.
- Yin D, Shumay E, Wang H-Y, Malbon CC (2005) Yeast Ste2 receptors as tools for study of mammalian protein kinases and adaptors involved in receptor trafficking. J. Mol. Signal 1: 2.
- Mcintyre N, Holdsworth CD, Turner DS (1964) New Interpretation Of Oral Glucose Tolerance. Lancet 2: 20–21. [crosref]
- Baggio LL, Drucker DJ (2007) Biology of incretins: GLP-1 and GIP. Gastroenterology 132: 2131–2157. [crosref]
- Alexander SPH, Benson HE, Faccenda E, Pawson, AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ (2013) CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br. J. Pharmacol. 170: 1459–1581.
- Nauck MA (2011) Incretin-based therapies for type 2 diabetes mellitus: properties, functions, and clinical implications. Am. J. Med. 2011 124: S3–S18.
- Quddusi S, Vahl TP, Hanson K, Prigeon RL, D‘Alessio A (2003) Differential effects of acute and extended infusions of glucagon-like peptide-1 on first- and second-phase insulin secretion in diabetic and nondiabetic humans. Diabetes Care 26: 791–798.
- Holst JJ, Seino Y (2009) GLP-1 receptor agonists: targeting both hyperglycaemia and disease processes in diabetes. Diabetes Res Clin Pract 85: 1–3. [crosref]
- Mentlein R, Gallwitz B, Schmidt WE (1993) Dipeptidyl-peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon-like peptide-1(7–36)amide, peptide histidine methionine and is responsible for their degradation in human serum. Eur. J. Biochem 214: 829–835.
- Scott DA, Boye KS, Timlin L, Clark JF, Best JH (2013) A network meta-analysis to compare glycaemic control in patients with type 2 diabetes treated with exenatide once weekly or liraglutide once daily in comparison with insulin glargine, exenatide twice daily or placebo. Diabetes Obes. Metab 15: 213–223.
- Butler AE, Campbell-Thompson M, Gurlo T, Dawson DW, Atkinson M, Butler PC (2013) Marked expansion of exocrine and endocrine pancreas with incretin therapy in humans with increased exocrine pancreas dysplasia and the potential for glucagon-producing neuroendocrine tumors. Diabetes 62: 2595–2604.
- Pabreja K, Mohd MA, Koole C, Wootten D, Furness SGB (2014) Molecular mechanisms underlying physiological and receptor pleiotropic effects mediated by GLP-1R activation. Br. J. Pharmacol. 171: 1114–1128.
- Nathan DM, Buse JB, Davidson, MB, Ferrannini E, Holman RR, Sherwin R, Zinman B (2009) American Diabetes Association and European Association for Study of Diabetes, Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 32: 193–203.
- SJ Dowell, AJ Brown (2002) Yeast assays for G-protein-coupled receptors, Recept. Channels 8: 343–352.
- Stewart GD, Valant C, Dowell SJ, Mijaljica D, Devenish RJ, Scammells PJ, Sexton PM, Christopoulos A (2009) Determination of adenosine A1 receptor agonist and antagonist pharmacology using Saccharomyces cerevisiae: implications for ligand screening and functional selectivity. J. Pharmacol. Exp. Ther 331: 271–286.
- Weston C, Poyner D, Patel V, Dowell S, Ladds G (2014) Investigating G protein signalling bias at the glucagon-like peptide-1 receptor in yeast. Br J Pharmacol 171: 3651–3665.[crosref]
- Montrose-Rafizadeh C, Avdonin P, MJ Garant, MJ Rodgers BD, Kole S, Yang H, Levine MA, Schwindiger S, Bernier M (1999) Pancreatic glucagon-like peptide-1 receptor couples to multiple G proteins and activates mitogen-activated protein kinase pathways in Chinese hamster ovary cells. Endocrinology 140: 1132–1140.
- Hallbrink M, Holmqvist T, Olsson M, Ostenson CG, Efendic S, Langel U (2001) Different domains in the third intracellular loop of the GLP-1 receptor are responsible for Galpha(s) and Galpha(i)/Galpha(o) activation. Biochim. Biophys. Acta 1546: 79–86.
- Coopman K, Huang Y, Johnston N, Bradley SJ, Wilkinson GF, Willars GB (2010) Comparative effects of the endogenous agonist glucagon-like peptide-1 (GLP-1)-(7–36) amide and the small-molecule ago-allosteric agent ‘compound 2’ at the GLP-1 receptor. J. Pharmacol. Exper. Therapeut. 334: 795–808.
- Brown AJ, Dyos SL, Whiteway MS, White JH, Watson MAE, Marzioch M, Clare JJ, Cousens DJ, Paddon C, Plumpton C, Romanos MA, Dowell SJ (2000) Functional coupling of mammalian receptors to the yeast mating pathway using novel yeast/mammalian G protein a-subunit chimeras. Yeast 16: 11–22.
- Dowell SJ, Brown AJ (2009) Yeast assays for G protein-coupled receptors. In: G Protein-Coupled Receptors in Drug Discovery 2009, New York, Humana Press 213–229.
- Ladds G, Davis K, Hillhouse EW, Davey J (2003) Modified yeast cells to investigate the coupling of G protein-coupled receptors to specific G proteins. Mol Microbiol 47: 781–792. [crosref]
- Ladds G, Goddard A, Davey J (2005) Functional analysis of heterologous GPCR signalling pathways in yeast. Trends Biotechnol 23: 367–373. [crosref]
- Miret JJ, Rakhilina L, Silverman L, Oehlen B (2002) Functional expression of heteromeric calcitonin gene-related peptide and adrenomedullin receptors in yeast. J. Biol. Chem. 27: 6881–6887.
- Dexter D, Moye-Rowley WS, Wu AL, Golin J (1994) Mutations in the yeast PDR3, PDR4, PDR7 and PDR9 pleiotropic (multiple) drug resistance loci affect the transcript level of an ATP binding cassette transporter gene, PDR5. Genetics 136: 505–515.
- Nourani A, Wesolowski-Louvel M, Delaveau T, Jacq C, Delahodde A (1997) Multiple-drug-resistance phenomenon in the yeast Saccharomyces cerevisiae: involvement of two hexose transporters. Mol. Cell. Biol.17: 5453–5460.
- Hallstrom TC, Moye-Rowley WS (1998) Divergent transcriptional control of multidrug resistance genes in Saccharomyces cerevisiae. J. Biol. Chem. 273: 2098–2104.
- Prendergast JA, Singer RA, Rowley N, Rowley A, Johnston GC, Danos M, Kennedy B, Gaber RF (1995) Mutations sensitizing yeast cells to the start inhibitor nalidixic acid. Yeast 11: 357–547.
- Arkinstall S, Payton M, Maundrel K (1995) Activation of phospholipase C gamma in Schizosaccharomyces pombe by coexpression of receptor or nonreceptor tyrosine kinases. Mol. Cell. Biol. 15: 1431–1438.
- Angermayr M, Strobel G, Müller G, Bandlow W (2000) Stable plasma membrane expression of the soluble domain of the human insulin receptor in yeast. FEBS Lett 481: 8–12.
- Müller G, Rouveyre N, Upshon C, Bandlow W (1998) Insulin signaling in the yeast Saccharomyces cerevisiae. 3. Induction of protein phosphorylation by human insulin. Biochemistry 37: 8705–8713.
- Moller NPH, Moller KB, Lammers R, Kharitonenkov A, Hoppe E, Wiberg FC, Sures I, Ullrich A (1995) Selective down-regulation of the insulin receptor signal by protein-tyrosine phosphatases alpha and epsilon. J. Biol. Chem. 270: 23126–23131.
- Yazici D, Sezer H (2017) Insulin Resistance, Obesity and Lipotoxicity. Adv Exp Med Biol 960: 277–304. [crosref]
- Rice Bradley BH (2018) Dietary Fat and Risk for Type 2 Diabetes: a Review of Recent Research. Curr Nutr Rep 7: 214–226. [crosref]
- Sabin MA, De Hora M, Holly JM, Hunt LP, Ford AL, Williams SR, Baker JS, Retallick CJ Crowne EC, Shield JP (2007) Fasting nonesterified fatty acid profiles in childhood and their relationship with adiposity, insulin sensitivity, and lipid levels. Pediatrics 120: 1426–1433.
- Kusminski CM, Shetty S, Orci L, Unger RH, Scherer PE (2009) Diabetes and apoptosis: lipotoxicity. Apoptosis 14: 1484–1495. [crosref]
- Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC (2008) Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab 10 Suppl 4: 32–42. [crosref]
- Lee J, Pilch PF (1994) The insulin receptor: structure, function, and signaling. Am J Physiol 266: C319–334. [crosref]
- Nolan CJ, Ruderman NB, Kahn SE, Pedersen O, Prentki M (2015) Insulin resistance as a physiological defense against metabolic stress: implications for the management of subsets of type 2 diabetes. Diabetes 64: 673–686.
- Smith U, Kahn BB (2016) Adipose tissue regulates insulin sensitivity: role of adipogenesis, de novo lipogenesis and novel lipids. J. Intern. Med. 280: 465–475.
- AN Onyango (2018) Cellular stresses and stress responses in the pathogenesis of insulin resistance. Oxid. Med. Cell. Longev 321714.
- Kahn SE, Hull RL, Utzschneider KM (2006) Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444: 840–846. [crosref]
- Pineau L, Ferreira T (2010) Lipid-induced ER stress in yeast and β cells: parallel trails to a common fate. FEMS Yeast Res 10: 1035–1045. [crosref]
- Ježek P, Jaburek M, Holendová B, Plecitá-Hlavatá L (2018) Fatty acid-stimulated secretion vs. lipotoxicity. Molecules 23: 1483.
- Morgan NG, Dhayal S, Diakogiannaki E, Welters HJ (2008) The cytoprotective actions of long-chain mono-unsaturated fatty acids in pancreatic I-cells. Biochem. Soc. Trans. 36: 905–908.
- Katsoulieris E, Mabley JG, Samai M, Green IC, Chatterjee PK (2009) (alpha)-Linolenic acid protects renal cells against palmitic acid lipotoxicity via inhibition of endoplasmic reticulum stress. Eur. J. Pharmacol 623: 107–112.
- Pineau L, Colas J, Dupont S, Beney L, Fleurat-Lessard P, Berjeaud JM, Berges T, Ferreira T (2009) Lipid-induced ER stress: synergistic effects of sterols and saturated fatty acids. Traffic 10: 673–690.
- Cunha DA, Hekerman P, L Ladriere L, Bazarra-Castro A, Ortis F, et al (2008) Initiation and execution of lipotoxic ER stress in pancreatic (beta)-cells. J. Cell Sci. 121: 2308–2318.
- Diakogiannaki E, Welters HJ, Morgan NG (2008)Differential regulation of the endoplasmic reticulum stress response in pancreatic (beta)-cells exposed to long-chain saturated and monounsaturated fatty acids. J. Endocrinol. 197: 553–563.
- Almanza A, Carlesso A, Chintha, C, Creedican S, Doultsinos D, et al (2018) Endoplasmic reticulum stress signalling – from basic mechanisms to clinical applications. FEBS J. 2018, doi: 10.1111/febs.14608, [Epub ahead of print].
- Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101: 249–258.
- Zhang YZ, Kaufman RJ (2006) The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology 66: S102-S109.
- Kincaid MM, Cooper AA (2007) ERADicate ER stress or die trying. Antioxid Redox Signal 9: 2373–2387. [crosref]
- Cox JS, Walter P (1996) A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 87: 391–404.
- Pineau L, Bonifait L, Berjeaud J.-M, Alimardani-Theuil P, Berges T, Ferreira T (2008) A lipid-mediated quality control process in the golgi apparatus in yeast. Mol. Biol. Cell 19: 807–821.
- Bódis K, Roden M (2018) Energy metabolism of white adipose tissue and insulin resistance in humans. Eur. J. Clin. Invest. 2018, e13017, [Epub ahead of print].
- McGarry JD (1992) What if Minkowski had been ageusic? An alternative angle on diabetes. Science 258: 766–770. [crosref]
- McGarry JD (2002) Banting lecture 2001: dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes 51: 7–18. [crosref]
- Cerk IK, Wechselberger L, Oberer M1 (2018) Adipose Triglyceride Lipase Regulation: An Overview. Curr Protein Pept Sci 19: 221–233. [crosref]
- Duncan RE, Ahmadian M, Jaworski K, Sarkadi-Nagy E, Sul HS (2007) Regulation of lipolysis in adipocytes. Annu Rev Nutr 27: 79–101. [crosref]
- Zechner R, Kienesberger PC, Haemmerle G, Zimmermann R, Lass A (2009) Adipose triglyceride lipase and the lipolytic catabolism of cellular fat stores. J. Lipid Res. 50: 3–21.
- Lafontan M, Langin D (2009) Lipolysis and lipid mobilization in human adipose tissue. Prog Lipid Res 48: 275–297. [crosref]
- Coleman RA, Mashek DG (2011) Mammalian triacylglycerol metabolism: synthesis, lipolysis, and signaling. Chem. Rev111: 6359–6386.
- Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, Gross RW (2004) Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J. Biol. Chem. 279: 48968–48975.
- Villena JA, Roy S, Sarkadi-Nagy E, Kim KH, Sul HS (2004) Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: ectopic expression of desnutrin increases triglyceride hydrolysis. J. Biol. Chem. 279: 47066–47075.
- R Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M, Lass A, Neuberger G, Eisenhaber F, Hermetter A, Zechner R (2004) Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 306: 1383–1386.
- Grönke S, Mildner A, Fellert S, Tennagels N, Petry S, Müller G, Jäckle H, Kühnlein RP (2005) Brummer lipase is an evolutionary conserved fat storage regulator in Drosophila. Cell Metabol 1: 323–330.
- Kurat CF, Natter K, Petschnigg J, Wolinski H, Scheuringer K, Scholz H, Zimmermann R, Leber R, Zechner R, Kohlwein SD (2006) Obese yeast: triglyceride lipolysis is functionally conserved from mammals to yeast. J. Biol. Chem. 281: 491–500.
- Chakrabarti P, Kim JY, Singh M, Shin Y-K, Kim J, Kumbrink J, Wu Y, Lee M-J, Kirsch KH, Fried SK, Kandror KV (2013) Insulin inhibits lipolysis in adipocytes via the evolutionarily conserved mTORC1-Egr1-ATGL-mediated pathway. Mol. Cell. Biol. 33: 3659–3666.
- Kitamura T, Kitamura Y, Kuroda S, Hino Y, Ando M, Kotani K, Konishi H, Matsuzaki H, Kikkawa U, Ogawa W, Kasuga M (1999) Insulin-induced phosphorylation and activation of cyclic nucleotide phosphodiesterase 3B by the serine-threonine Akt. Mol. Cell. Biol. 19: 6286–6296.
- Choi SM, Tucker DF, Gross DN, Easton RM, DiPilato LM, Dean AS, Monks BR, Birnbaum MJ (2010) Insulin regulates adipocyte lipolysis via an Akt-independent signaling pathway. Mol. Cell. Biol. 30: 5009–5020.
- Chakrabarti P, English T, Shi J, Smas CM, Kandror KV (2010) The mTOR complex 1 suppresses lipolysis, stimulates lipogenesis and promotes fat storage. Diabetes 59: 775–781.
- Grönke S, Müller G, Hirsch J, Fellert S, Andreou A, et al. (2007) Dual lipolytic control of body fat storage and mobilization in Drosophila. PLoS Biol 5: e137. [crosref]
- Luong N, Davies CR, Wessells RJ, Graham SM, King MT, et al. (2006) Activated FOXO-mediated insulin resistance is blocked by reduction of TOR activity. Cell Metab 4: 133–142. [crosref]
- Birse RT, Choi J, Reardon K, Rodriguez J, Graham S, et al. (2010) High-fat-diet-induced obesity and heart dysfunction are regulated by the TOR pathway in Drosophila. Cell Metab 12: 533–544. [crosref]
- Fernandez-Almonacid R, Rosen OM (1987) Structure and ligand specificity of the Drosophila melanogaster insulin receptor. Mol. Cell. Biol. 7: 2718–2727.
- Boyle KB, Hadaschik D, Virtue S, Cawthorn WP, Ridley SH, O‘Rahilly S, Siddle K (2009) The transcription factors Egr1 and Egr2 have opposing influences on adipocyte differentiation. Cell Death Diff. 16: 782–789.
- Wang W, Huang L, Huang, Y, Yin JW. Berk AJ, Friedman JM, Wang G (2009) Mediator MED23 links insulin signaling to the adipogenesis transcription cascade. Dev. Cell 16: 764–771.
- Brand F, Herrmann SM, Nicaud V, Evans A, Ruidavets JB, Arveiler D, Luc G, Cambien F, Soubrier F (2000) Identification of two polymorphisms in the early growth response protein-1 gene: possible association with lipid variables. J. Mol. Med. 78: 81–86.
- Gokey NG, Lopez-Anido C, Gillian-Daniel AL, Svaren J (2011) Early growth response 1 (egr1) regulates cholesterol biosynthetic gene expression. J. Biol. Chem 286: 29501–29510.
- Shen N, Yu X, Pan FY, Gao X, Xue B, Li CJ (2011) An early response transcription factor, egr-1, enhances insulin resistance in type 2 diabetes with chronic hyperinsulinism. J. Biol. Chem. 286: 14508–14515.
- Estruch F, Carlson M (1993) Two homologous zinc finger genes identified by multicopy suppression in a SNF1 protein kinase mutant of Saccharomyces cerevisiae. Mol. Cell. Biol. 13: 3872–3881.
- Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, et al. (1996) Life with 6000 genes. Science 274: 546, 563–567. [crosref]
- Blattner FR, Plunkett G 3rd, Bloch CA, Perna NT, Burland V, et al. (1997) The complete genome sequence of Escherichia coli K-12. Science 277: 1453–1462. [crosref]
- Wolin SL (1994) From the elephant to E. coli: SRP-dependent protein targeting. Cell 77: 787–790. [crosref]
- King K, Dohlman HG, Thorner J, Caron MG, Lefkowitz R (1995) Control of yeast mating signal transduction by a mammalian ß2-adrenergic receptor and Gs a subunit. Science 250: 121–123.
- Dohlman HG, Apaniesk D, Chen Y, Song J, Nusskern D (1995) Inhibition of G-protein signaling by dominant gain-of-function mutations in Sst2p, a pheromone desensitization factor in Saccharomyces cerevisiae. Mol. Cell. Biol. 15: 3635–3643.
- Proce LA, Kajkowski EM, Hadcock JR, Ozenberger BA, Pausch MH (1995) Functional coupling of a mammalian somatostatin receptor to the yeast pheromone response pathway. Mol. Cell. Biol. 15: 6188–6195.
- Pausch MH (1997) G-protein coupled receptors in Saccharomyces cerevisiae: high throughput screening assays for drug discovery. Trends Biotechnol. 15: 487–494.
- Klein C, Paul JI, Sauvé K, Schmidt MM, Arcangeli L, et al. (1998) Identification of surrogate agonists for the human FPRL-1 receptor by autocrine selection in yeast. Nat Biotechnol 16: 1334–1337. [crosref]
- Vedovato N, Ashcroft FM, Puljung MC (2015) The Nucleotide-Binding Sites of SUR1: A Mechanistic Model. Biophys J 109: 2452–2460. [crosref]
- Benarroch EE (2017) Sulfonylurea receptor-associated channels: Involvement in disease and therapeutic implications. Neurology 88: 314–321.
- Hahnenberger KM, Krystal M, Esposito K, Tang W, Kurtz S (1996) Use of microphysiometry for analysis of heterologous ion channels expressed in yeast. Nat Biotechnol 14: 880–883. [crosref]
- Bertin B, Freissmuth M, Jockers R, Strosberg AD, Marullo S (1994) Cellular signaling by an agonist-activated receptor/Gs alpha fusion protein. Proc Natl Acad Sci U S A 91: 8827–8831. [crosref]
- Hall BL, Smit-McBride Z, Provalsky ML (1993) Reconstitution of retinoid X receptor function and combinatorial regulation of other nuclear hormone receptors in the yeast Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 90: 6929–6933.
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, et al. (1995) The nuclear receptor superfamily: the second decade. Cell 83: 835–839. [crosref]
- Chambon P (1996) A decade of molecular biology of retinoic acid receptors. FASEB J 10: 940–954. [crosref]
- Supert-Furga, G.; Jonasson, K.; Courtneidge, S.A. A functional screen in yeast for regulators and antagonizers of heterologous protein tyrosine kinases. Nature Biotechnol. 1996, 14, 600–604.
- Fields S, Song OK (1989) A novel genetic system to detect protein-protein interactions. Nature 340: 245–246.
- Germino FJ, Wang ZX, Weissman SM (1993) Screening for in vivo protein-protein interactions. Proc Natl Acad Sci U S A 90: 933–937. [crosref]
- Vidal M, Brachmann RK, Fattaey A, Harlow E, Boeke JD (1996) Reverse two-hybrid and one-hybrid systems to detect dissociation of protein-protein and DNA-protein interactions. Proc. Natl. Acad. Sci. USA 93: 10315–10320.
- Ozenberger BA, Young KH (1995) Functional interaction of ligands and receptors of the hematopoietic superfamily in yeast. Mol Endocrinol 9: 1321–1329. [crosref]
- Kajkowski EM, Proce LA, Paush MH, Young KH, Ozenberger BA (1997) Investigation of growth hormone releasing hormone receptor structure and activity using yeast expression technologies. J. Recept. Signal Transduct. Res. 17: 293–303.
- Zhu J, Kahn CR (1997) Analysis of a peptide hormone-receptor interaction in the yeast two-hybrid system. Proc. Natl. Acad. Sci. USA 94: 13063–3068.
- Tirode F, Malaguti C, Romero F, Attar R, Camonis J, et al. (1997) A conditionally expressed third partner stabilizes or prevents the formation of a transcriptional activator in a three-hybrid system. J Biol Chem 272: 22995–22999. [crosref]
- SenGupta DJ, Zhang B, Kraemer B, Pochart P, Fields S, et al. (1996) A three-hybrid system to detect RNA-protein interactions in vivo. Proc Natl Acad Sci U S A 93: 8496–8501. [crosref]
- Lieberles S, Diver ST, Austin DJ, Schreiber SL (1997) Inducible gene expression and protein translocation using nontoxic ligands identified by a mammalian three-hybrid screen. Proc. Natl. Acad. Sci. USA 94: 7825–7830.
- Catterall WA (1998) Yeasty brew yields novel calcium channel inhibitor. Nat Biotechnol 16: 906. [crosref]
- Young K, Stephen L, Sun L, Lee E, Modi M, Hellings S, Husbands M, Ozenberger B, Franco R (1998) Identification of a calcium channel modulator using a high throughput yeast two-hybrid screen. Nature Biotechnol 16: 946–950.
- Leach M (1994) Discovery on a credit card? Drug Discov. Today 2: 263–254.
- Sedlak BJ (1997) Gene chip technology ready to impact diagnostic markers. Gen. Eng. News 17: 1, 13, 34.
- Chiem NH, Harrison DJ (1998) Microchip systems for immunoassay: an integrated immunoreactor with electrophoretic separation for serum theophylline. Clin. Chem. 44: 591–598.
- Uhlig A, Lindner E, Teutloff C, Schnakenberg U, Hintsche R (1997) Miniaturized ion-selective chip electrode for sensor application. Anal Chem 69: 4032–4038. [crosref]
- Liu CC, Neuman MR, Romankiw LT, Makovos EB (1987) Microelectronic sensors for simultaneous measurement of PO2 and pH. Adv Exp Med Biol 220: 295–298. [crosref]
- Huang J, Schreiber SL (1997) A yeast genetic system for selecting small molecule inhibitors of protein-protein interactions in nanodroplets. Proc. Natl. Acad. Sci. USA 94: 13396–13401.