DOI: 10.31038/CST.2018341
Abstract
Cancer remains as one of the most challenging diseases to treat. However, this era has commenced with the introduction of novel drug treatments that are safer, and less toxic. The efficacy of a novel metabolic therapy, dichloroacetate sodium (DCA) was investigated.
27 solid-tumors were studied; 3 of 27 exhibited high or intermediate sensitivity to DCA as a single agent; 7 of 27 exhibited high or intermediate sensitivity to DCA in combination with chemotherapeutic agent(s). 9 of 27 exhibited no sensitivity to DCA as a single agent or in combination.
Clinical outcomes further validated the in vitro data.
Our findings indicate a potential role for DCA in oncology therapeutics in a wide range of cancer types. However, the diversity of the tumor responses among organ-specific cancer types underscores the necessity to conduct clinical studies on an individual basis rather than with a “one-size-fits-all” approach. The relative clinical safety, well- characterized pharmacodynamic profile, low side effects, and low cost of DCA further makes it an ideal candidate for development as an effective anticancer agent. Ideally, randomized controlled clinical trials should be designed to further correlate and validate this preliminary pilot study in the oncology setting and to fully appreciate the impact of DCA on cancer recurrence, response rates and survival rates.
Key Words
DCA, mitochondria, solid-tumors, oncology, chemotherapy, Warburg Effect
Introduction
Cytotoxic chemotherapeutic treatment regimens tend to be deleterious and toxic to cancer patients. Furthermore, these treatments often come with significant trade-offs: treatment may have to be halted because of cumulative toxicity; treatment may produce long-term complications; or the drug(s) that kill the cancer may permanently damage healthy organs [1] , or worse. Thus, today many clinicians are changing their clinical practices by opting for targeted and/or ancillary drug treatments that kill the tumor cell populations while sparing healthy cells, thus affording the patient a valuable quality-of-life.
It is known that greater than 70% of all cancer types rely on aerobic glycolysis for energy production, which is an inefficient means of generating ATP, a feature that becomes an advantageous biomarker. Aerobic glycolysis is a result of malfunctioning/hyperpolarized mitochondria. Cancer cells generally express increased aerobic glycolysis in the cytosol (Warburg Effect/lactic acid fermentation) [2] rather than oxidative phosphorylation (normal cells) for energy production,[3] thus producing excessive lactate and therefore inducing a low pH microenvironment [4].
In 2007, Drs. Archer and Michelakis from the University of Alberta, Canada, [5] decreed the use of Dichloroacetate (DCA) as a general use metabolic chemotherapeutic agent that could reverse this mitochondrial hyperpolarized state thus inducing cancer cells to undergo apoptosis.
The ability of DCA to decrease lactate production has been used for more than 30 years in the treatment of lactic acidosis in inherited mitochondrial diseases in humans [6] Lactic acidosis is a physiological condition characterized by low pH in the body tissues and blood accompanied by the buildup of lactate [7]. The condition typically occurs when cells become hypoxic thus impairing cellular respiration leading to the lower pH levels (acidosis). Simultaneously, cells are forced to metabolize glucose anaerobically, which leads to lactate formation. Therefore, elevated lactate is indicative of tissue hypoxia, hypoperfusion, manifesting in possible tissue damage [8]. The characteristics of mitochondrial diseases in humans are virtually identical to tumorigenesis, complete with the inefficient bioenergetic mitochondria. This property has led to trials of DCA for the treatment in humans presenting with a variety of cancers [9].
The generic drug sodium dichloroacetate (DCA) is an orally bioavailable small molecule that, by inhibiting pyruvate dehydrogenase kinase (PDK), increases the flux of pyruvate into the mitochondria, promoting glucose oxidation. This reverses the suppressed mitochondrial apoptosis in cancer cells and results in suppression of tumor growth in vitro and in vivo [6]. Thus, it would be reasonable to propose that cells with mitochondrial defects, or cells in a high glycolytic and hypoxic environment would likely be more sensitive to glycolytic inhibition by DCA. Therefore, a prospective study of the efficacy of DCA as a potential chemotherapeutic agent was conducted.
Materials and Methods
A variety of fresh solid tumor specimens (27) were procured from patients of a private clinic, Medicor Cancer Centres Inc. (Toronto, Ontario, Canada) The tumor specimens were either obtained from biopsies of superficial metastases, superficial lymph nodes infiltrated with metastases, or at the time of major cancer surgery. The tumor specimens were accredited by the attending pathologist to be comprised of tumor tissue. Patients were provided with a written informed consent to perform the CS/CR (chemosensitivity / chemoresistance) assay. The live tumor samples obtained were then mechanically disaggregated to obtain single-cell heterogenates (SCH). The SCH were then incubated at 360C / 5% CO2 for 48 hours in a humidified chamber to allow for equilibration. Following incubation, the SCH were washed, counted, and a small aliquot stained with trypan blue, to assess initial viability. Twenty thousand cells were added per analysis tube. The chemotherapeutic agents (obtained from Sigma-Aldrich; Selleck Chemical, and McKesson) were added at peak plasma concentrations (Cmax), plus/minus DCA (at peak plasma concentration/Cmax), and incubated at 360C / 5% CO2 for 72 hours in a humidified chamber. After 72 hours, the SCH were washed and tagged with green fluorescein LIVE/DEAD® Fixable Stains for Flow Cytometry (Molecular Probes). The reactive dye can permeate the compromised membranes of dead cells and react with free amines on the interior and exterior of the cell, whereas only membrane-exterior free amines of viable cells are available to react with the dye, resulting in intense or dim staining, respectively. SCH in vitro chemotherapy response was determined using a Becton Dickinson FACScan flow cytometer* and SCH analyzed for percentage of live versus dead cell populations against a live non-drug control. A dead cell control was also used consisting of SCH placed at 560C for 1 hour.
*All specimens were high grade / metastatic tumors unless noted; no tumor was naïve; no tumor was a primary 10,000 events were counted for each SCH aliquot.
Results:
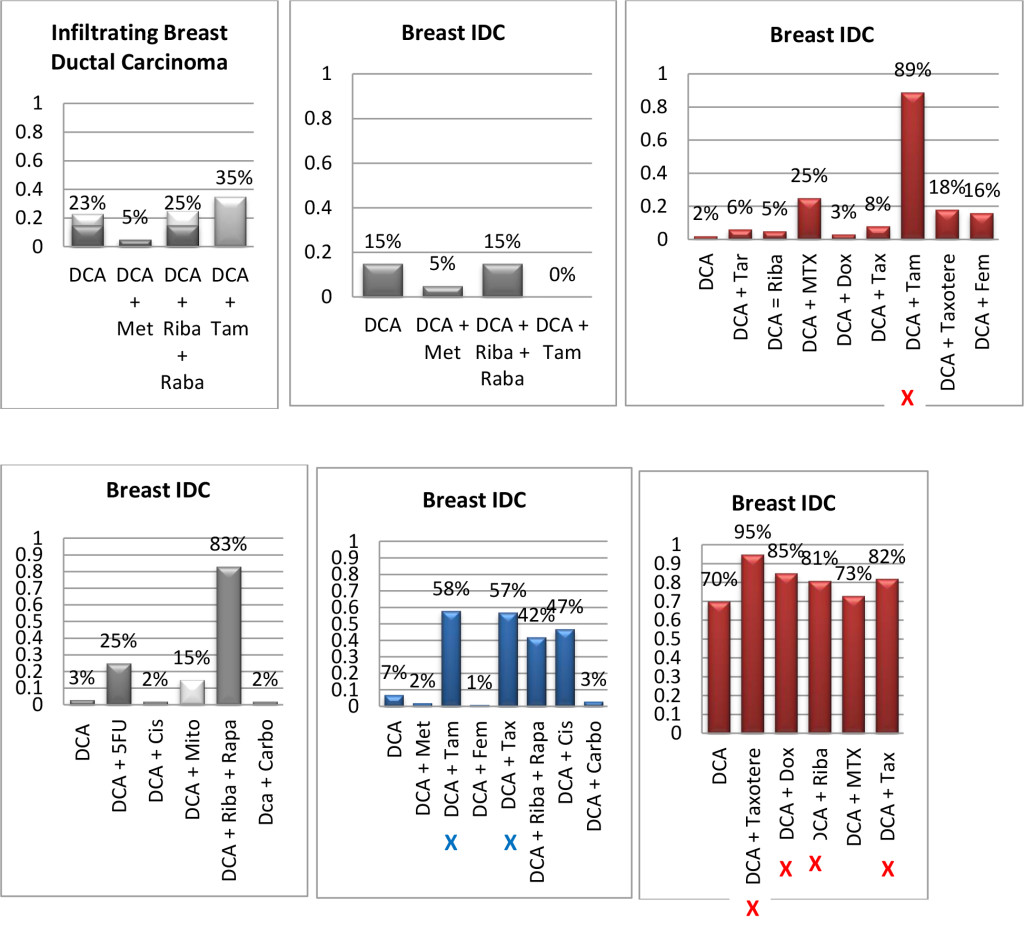
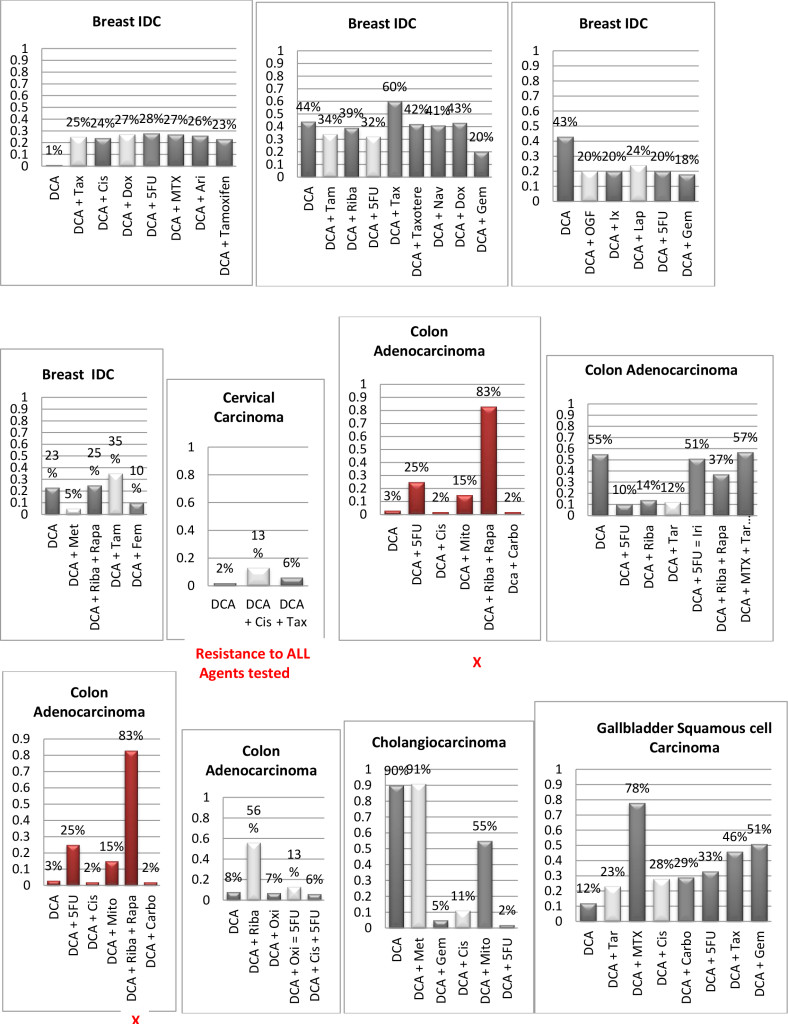
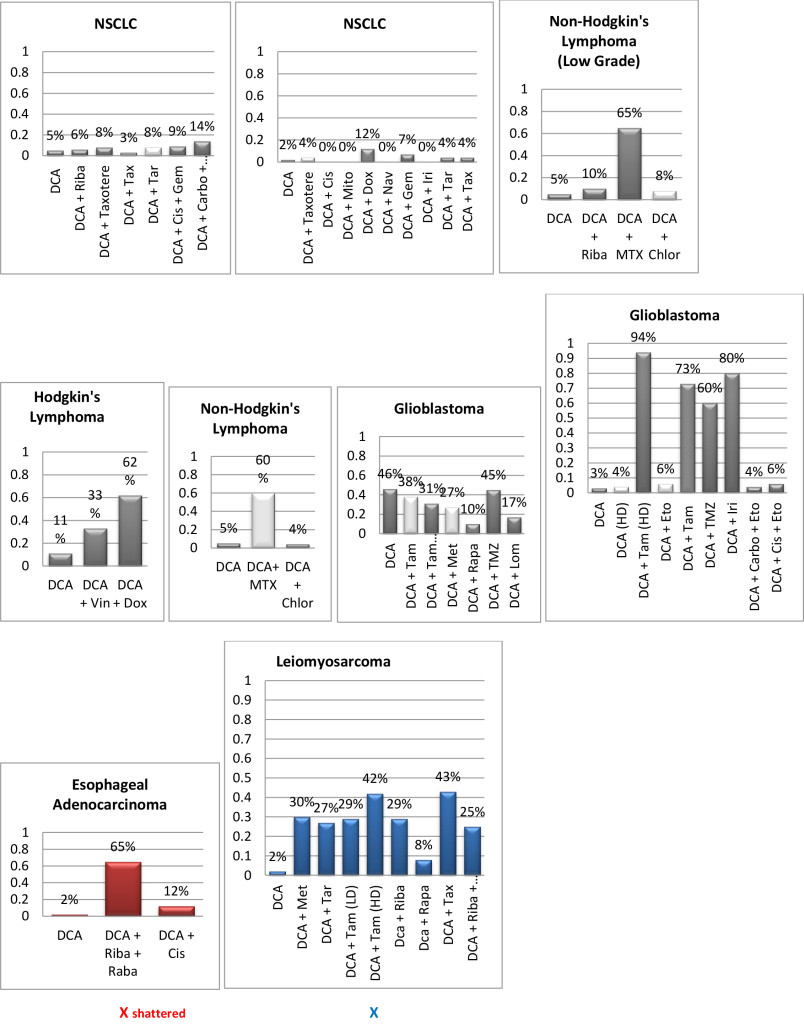
Figure 1. Histograms/Graphs
Unless Noted: DCA inhibited the conventional therapeutic drug; or no synergy was noted with the conventional therapeutic drug; or if synergy was LDS; or inhibition of both agents when combined. This is noted by the Dark Colored Histograms.
Red Colored Histograms (X); Synergy (HDS) when conventional chemotherapeutic drug was combined with DCA
Blue Colored Histograms (X); Synergy (IDS) when conventional chemotherapeutic drug was combined with DCA
Note: 1) In our assay if percent kill was not > 33%, treatment was designated as LDS (Low Drug Sensitivity) and as such was not considered an efficacious treatment option; 34%-65% kill was designated as IDS (Intermediate Drug Sensitivity) a partial response may be obtained; > 66% kill was designated as HDS (High Drug Sensitivity) and a favorable response could be expected.
2) Definitions: Permissive: drug as a single agent is non-effective unless in combination with another agent; Additive: in combination the drugs produce a total effect the same as the sum of the individual effects; Synergy: in combination the drugs produce a total effect that enhance or magnify the sum of the individual effects; Inhibition: in combination the drugs produce a total effect that inhibits the sum of the efficacy of the effective drug(s).
Table 1. SAMPLE CLINICAL RESULTS: Previously Un-Published and Unrelated to Figure 1 Data
|
Total-27 solid-Tumors Sensitivity to DCA |
|||
|
Single Agent |
Efficacy |
Combination |
Efficacy |
|
11% |
HDS |
15% |
HDS |
|
15% |
IDS |
22% |
IDS |
|
33% |
LDS/NONE |
33% |
LDS/NONE |
|
11/27 Breast solid-Tumors Sensitivity to DCA |
|||
|
Single Agent |
Efficacy |
Combination |
Efficacy |
|
24% |
HDS |
15% |
HDS |
|
6% |
IDS |
24% |
IDS |
|
33% |
LDS/NONE |
33% |
LDS/NONE |
|
4/27 Colon solid-Tumors Sensitivity to DCA |
|||
|
Single Agent |
Efficacy |
Combination |
Efficacy |
|
0% |
HDS |
25% |
HDS |
|
25% |
IDS |
50% |
IDS |
|
0% |
LDS/NONE |
0% |
LDS/NONE |
- 32 year old male, leg melanoma, treated with wide excision and inguinal node dissection, local recurrence and progressive inguinal lymphadenopathy post-op while receiving natural therapy only, CT proven complete response to oral DCA therapy for over 3 yearswith no concurrent conventional therapies.
- 63 year old female, non-Hodgkins lymphoma treated with standard chemotherapy, marrow injury from chemo (stopped), progression while off treatment, CT-proven stable disease for 2 years while taking oral DCA and no concurrent conventional therapy.
- 80 year old male with transitional cell bladder carcinoma, recurrent disease after multiple resections and BCG, cystoscopy- proven tumour shrinkage with short course of oral DCA (6 weeks), re-treated after 1 year, delayed radical cystectomy for 4 years.
- 31 year old female with frontotemporal grade 3 astrocytoma transformed to glioblastoma, treated with debulking surgery followed by chemoradiation. Patient received DCA for 3 months following chemoradiation, with no concurrent chemotherapy, and no subsequent conventional therapy. Initial MRI appeared to show rapid progression with patient remaining asymptomatic. MRI deemed to reflect pseudoprogression. Patient had a complete response and remains alive and well 6 years post-treatment.
- 67 year old female with recurrent transitional cell bladder carcinoma following multiple TURBT procedures and intravesical chemotherapy. Treated with oral DCA 26mg/kg/day for 6 weeks on a cycle of 2 weeks on and 1 week off. DCA stopped due to neuropathy. Disappearance of recurrent solitary bladder tumour by pelvic ultrasound, confirmed by cystoscopy and repeated negative urine cytology reports. Patient remained clear at 6 months post-DCA therapy. Started low dose naltrexone combined with purified honokiol (magnolia extract) for recurrence prevention. Remains clear of bladder cancer 3 years following therapy.
Results/Discussion
Early carcinogenesis occurs in a hypoxic microenvironment and thus the transformed cells initially rely on aerobic glycolysis for energy production [4]. However, this early metabolic adaptation appears to also offer a proliferative advantage, suppressing apoptosis. Furthermore, the byproducts of glycolysis (i.e. lactate and acidosis) contribute to the breakdown of the extracellular matrix, facilitate cell mobility, and increase the metastatic potential [11]. Moreover, even though the tumors eventually become vascularized and O2 levels increase, the glycolytic phenotype persists, resulting in the ‘‘paradox’’ of glycolysis during aerobic conditions, the Warburg effect [2].
Aerobic glycolysis is a common metabolic alteration of tumor cells that results in overt lactic acid production, adapting the cells to tumor microenvironments and is necessary for their survival. Although lactate production results in less ATP per molecule of glucose, it has been shown that increased glycolysis and decreased oxidative phosphorylation may serve to increase the rate of ATP production without producing reactive oxygen species [2]. Indeed tumor cells do not suffer from ATP deficiency; in fact they generate more energy than normal cells, by increasing the level of glycolysis several-fold to support their enhanced growth and proliferation.12 It has also been shown that the Warburg effect is also involved in the avoidance of apoptosis [2]. Alternatively and paradoxically, the Warburg effect might serve to increase the biomass to provide nucleotides and lipid material necessary for rapidly dividing cells [13]. This theory is supported by the fact that signaling pathways such as AKT/mTOR, are known to play a role in biomass production, which also control aspects of the Warburg effect [13].
Moreover, it is well established that solid tumors tend to have a more acidic microenvironment than normal tissues [2]. Intracellular acidic water holds very little oxygen while an alkaline water micromilieu can hold large amounts of oxygen. It follows, then, that the more acidic the tumor cells, the less intracellular oxygen will be available. Thus this acidic phenotype would further support enhanced proliferation and hence tumorigenesis [15]. Indeed, it has been reported that due to this acidic milieu, an unusual reprogramming phenomenon will be the fate of some somatic cells. They can be drastically altered through changes and committed to a specific lineage and thus converted into a pluripotent state (capable of differentiating into nearly all cell types) when exposed to an environmental stress, in this case short exposure to low pH. This reprogramming process does not need nuclear manipulation or the introduction of transcription factors, thought to be necessary to induce pluripotency. This research group calls the phenomenon “stimulus-triggered acquisition of pluripotency” (STAP) [16].
Further support for tumors utilizing this bioenergetic inefficient, non-mitochondrial means of generating ATP has been shown by tumor cells exclusive expression of the embryonic M2 isoform of pyruvate kinase M2 which is necessary for aerobic glycolysis [14]. This unique phenotype provides a selective growth advantage for tumor cells in vivo and is associated with suppression of mitochondrial function and thus resistance to apoptosis, a further hallmark that characterizes cancer.
The Parra-Bonilla group demonstrate that pulmonary artery microvessel endothelial cells preferentially utilize glycolysis to generate ATP (Warburg effect), which may be necessary to sustain their growth and other rapidly growing untransformed cells [17]. Others have also demonstrated that AKT (Protein Kinase B, a serine/threonine-specific protein kinase that plays a key role in multiple cellular processes such as glucose metabolism, apoptosis, cell proliferation, transcription and cell migration). is activated by latent Kaposi’s sarcoma-associated herpes virus (KSHV) infection of endothelial cells [18,19]. KSHV infection of endothelial cells also activates hypoxia-induced factors HIF -1 and HIF-2 [19]. Further, AKT and HIFs have been shown to play prominent roles in the Warburg effect. During latent infection of endothelial cells, KSHV induces aerobic glycolysis and lactic acid production while decreasing oxygen consumption, leading to endothelial cell activation and thus angiogenesis promotion via the hypoxic milieu [20].
Lactic acidosis is characterized by tissue lactate levels of >5 mmol/L concurrently with serum levels of pH <7.35. [21]. Researchers at the University of Regensburg, Germany [22] show that intratumoral concentrations of lactic acid vary by tumor type as well as from tumor burden. They collected serum of 160 patients suffering from different malignancies and determined that patients with high tumor burden indeed present with a significant increase in serum lactate levels. Furthermore, since a characteristic feature of the tumor environment is local acidosis, they investigate the direct effect of lactic acid on T-cell proliferation, showing lactic acid inhibits T-cell proliferation as well as an intracellular increase concentration of lactic acid in the T-cell itself of10–20 mmol/L [22].
Taken in totality, it appears that virtually all cells associated with the tumor microenvironment play prominent roles in the Warburg effect.
But, Michelakis et al, demonstrate that this metabolic-electrical remodeling is an adaptive response and thus reversible. Since cancer cells are relatively deficient in Kv channels, [5,11] one could reverse the suppression of PDC (pyruvate dehydrogenase complex) activity, and thus increase apoptosis. The metabolic and the apoptotic pathways converge in the mitochondria and thus not independent from each other and therefore the glycolytic phenotype is associated with a state of apoptosis resistance [23].
Many glycolytic enzymes have been recognized to also regulate apoptosis, and several oncoproteins also induce the expression of glycolytic enzymes [24]. For example, AKT, which stimulates glycolysis and induces resistance to apoptosis, activates hexokinase, an enzyme catalyzing the first and irreversible step in glycolysis [25]. Via its downstream mediator glycogen synthase kinase 3 (GSK3), AKT induces the translocation of hexokinase to the mitochondrial membrane where it binds to the voltage-dependent anion channel (VDAC), suppressing apoptosis [25]. Inhibition of GSK3 in cancer cells causes unbinding of hexokinase from VDAC, induces apoptosis, and increases sensitivity to chemotherapyv [26].
DCA enters the cancer cell switching cancer promoting/inhibiting genes on or off including mtDNA. However, it appears that DCA requires an ectopic membrane transporter protein called SLC5A8 to enter the cancer cells. SLC5A8 mediates acetate transport in a Na+-coupled manner, with the affinity of dichloroacetate for the transporter ~45-fold higher than that of Na+, (dichloroaceate/ Na+ stoichiometry for the transport process is 2: 1.) [27]. When it does so, it restores mitochondrial function by reversing the ionic remodeling of hyperpolarized mitochondria, thus restoring apoptosis, allowing cancer cells to commit “suicide” which results in tumor shrinkage. Indeed, it has been shown that DCA does have broad spectrum anticancer properties with minimal toxicity in animal models, and has efficacy in humans including the treatment of glioblastoma (by virtue of its ability to cross the blood-brain barrier). DCA causes depolarization of mitochondria in GBM tissue but not in healthy brain tissue, as this tissue possesses ectopic expression of the SLC5A8 transporter [28].
Several studies have shown that DCA induces apoptosis, in a variety of cancer cell lines and as the mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth [29–31]. However, a recent investigation was not able to confirm these findings [32]. In correlation with our pilot studies we also observed that even though DCA was able to induce mitochondrial depolarization (Figure 2), we observed highly variable induction of apoptosis or necrosis when DCA was used as a single agent, or even as a chemosensitizer (Figure 1). Nonetheless, long and continuous in vivo exposure may be required as demonstrated by Bradford and Khan [33] and/or DCA may cause cell growth inhibition without causing apoptosis [34] and hence account for minimal in vitro results noted in the third decade (apoptosis) and thus account for the clinical ‘stable disease’ case noted above(as well as other unpublished cases observed at Medicor Cancer Centres)
Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo [35]. This would not be detected by the ChemoFit assay. We show that DCA selectively targets cells with defects in the mitochondrial and could demonstrate apoptosis or necrosis when DCA was combined with conventional chemotherapies thus acting as a chemosensitizer inducing synergistic effects on various tumor types. Moreover, Stockwin et al [32] demonstrate that a very high concentration of the compound (≥25 mM) was required to induce apoptosis, wherein our studies incorporated peak plasma concentrations as well as exposure time of the SCH to DCA was 72 hours and not beyond. A limiting factor in the study is the use of “fresh” tumor cells (not cell lines) and thus the inability to use cultures for extended periods of time, which would be required to measure growth inhibition.
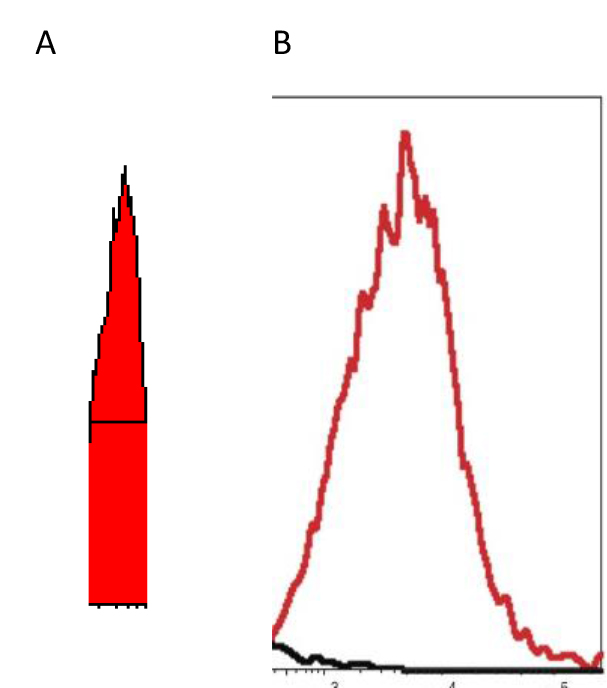
Figure 2. “X” scale – logarithm – fluorescence intensity spanning four decades (a 10,000-fold range)
“Y” scale – logarithm – cell Number
EXAMPLE-When live tumor cells are run through the flow cytometer without any drugs added, the histogram exhibits a peak in the 100–102 as noted in “A” the “ghost” peak; whereas if DCA is added to the aliquot of live tumor cells, the peak becomes very narrow, less ‘choppy’, and falls square in the middle of the 2 decades, as noted in “B” the “solid” peak– indicating reversing of the hyperpolarized mitochondria – the cell populations are “healthier?” [Unpublished Data]
A 1982 and a 1988 paper by Chen, et al. show that rhodamine 123 accumulates by various cancers and normal cells. The rhodamine 123 molecule, carries a net positive charge, and as such is accumulated and retained in areas of the cell that are more negatively charged in greater amounts and for longer periods of time than in less negatively charged areas [34–35]. Thus, retention of Rh123 in the mitochondria of many carcinomas suggests that the mitochondria in such cells are hyperpolarized. Due to this biochemical property, Chen points to two types of cancer that do not retain Rh123, sarcoma and oat cell lung cancer (SCLC). The 1988 paper also mentions as exceptions “large cell carcinomas of the lung” and “poorly differentiated carcinoma of the colon.” This is not definitive since there is certainly much variation among all types of cancer cells, but in light of the data contained in the Chen papers, and given the importance of the normalization of mitochondrial membrane potential to the apoptosis-inducing mechanism described by Michelakis, [36] it seems reasonable to assume that sarcomas, and small cell lung cancers are unlikely to respond to DCA and perhaps partially explain the results of our current study. However, in clinical practice of using DCA for over 7 years, Khan has observed both sarcoma and small cell lung cancers respond well (unpublished data), again highlighting the variability of individual tumor behaviors and the need to individualize therapy. Although neurotoxicity is a known and rather common side-effect and was indeed noted in the patients, it was reversible upon withdrawal of the drug or when treated with natural neuroregenerative medicinessuch as lipoic acid and B vitamins
Many of the patients who supplied tumours listed in Fig 1 could not be followed to determine if the in vivo responses matched the in vitro results noted above. The reasons were:
- the patient’s condition changed, and they were unable to take chemotherapy,
- the patient’s oncologist refused to prescribe the assay-guided therapy,
- the patient was lost to follow-up.
Since DCA had been used for years to treat rare metabolic disorders and was known to be relatively safe, [6] the potential existed for rapid translation of these findings to clinical use in the oncology setting. However, our pilot studies using DCA to restore normal generation of ATP and therefore reverse the resistant apoptosis phenotype, show that most of the tumors did not respond to DCA as a single agent or in combination with conventional agents. 3 of 10 breast cancer subtypes had intermediate or high sensitivity to DCA as a single agent, DCA also exhibited high efficacy when combined with various chemotherapeutic agents in the same 3 breast tumors. We noted 1 of 4 colon cancer subtypes had intermediate sensitivity to DCA as a single agent, and the only bile duct cancer tested had high sensitivity to DCA as a single agent.
Actually our data indicates that DCA inhibited the sensitivity of many of the conventional agents used including those used on breast and brain tissue that we hypothesized would be effective as noted above by other research groups. It should also be noted that although we analyzed “fresh” tumor tissue and any components associated with the micromilieu, many research groups tested DCA on human cells cultured outside the body and found that it killed lung, breast and brain cancer cells, but not healthy cells [29–32]. The issue of using fresh tissue versus cells lines; cells in cultures always present with concern and relevance. Cell lines are homogeneous rather than representing the heterogenic milieu of a specific patient’s tumor mass. As such, results for a given therapeutic agent(s) may not represent the individual‘s specific response and actually may reflect false positive or false negative effects. Further, allowing cells to proliferate in vitro does not represent the original tumor mass and thus not reflect in vivo response dynamics.
As mentioned, not all of the tumors responded to DCA as a single agent or in combination with conventional agents. There are several possible explanations for this. It is possible that the resistant tumors do not express the membrane transporter protein SLC5A8. It is known to be silenced in many tumors and not ectopic, which has been shown to be required for DCA entry into the cancer cells [28]. The tumor specimens analyzed were high grade / metastatic tumors and hence had prior exposure, if not multiple exposures, to drugs and radiation prior to analyses. It is also possible that the tumors had developed cross resistance to DCA as a result of prior treatment with multiple cytotoxic agents. It has also been shown that when the tumor bulk has not been effectively eradicated, the risk of recurrence and metastasis is high, [37] hence, the efficacy of DCA may be higher when it is administered in patients with low tumor burden. Thus, as mentioned above, most of our patient population was of high tumor burden explaining our subdued results. Another conjecture is that certain tumors may be able to utilize alternate fuels to generate ATP when glycolysis is shut down by DCA (e.g. ketone bodies or free fatty acids). Moreover, a limitation of the assay is based on altered cell membrane permeability but the initial site of cellular damage caused by some toxic agents is intracellular. Therefore, cells may be irreversibly damaged and committed to die, while the plasma membrane is still intact. Thus this assay could underestimate cell death. Despite this fact, these types of assays are widely used, accepted and correlate with clinical outcomes [38]. Finally, if DCA is cytostatic (growth inhibition without apoptosis) as shown by Blackburn’s group, [36] instead of cytolytic/cytotoxic, a cell death assay will not detect this influence [37, 39, 40].
Conclusion
Despite the challenges that remain in treating cancer, this era has commenced with the introduction of novel drug treatments that are safer, and less toxic. Thus, many clinicians are changing their clinical practices by opting for these “gentler” “targeted” treatments that kill the tumor cells and leave normal cells unaffected. Furthermore, it appears that with the advent of targeted therapies, and the prediction that individualizing therapy is now an appropriate method for treating cancer, many physicians are now realizing the value of CS/CR testing, and advocating its use to guide them in choosing a chemotherapeutic regimen. Microscopic histological “sameness” does not equate to tumor genetic, epigenetic and phenotypic “sameness”. Indeed, the characteristics and behavior of specific cancer types differs widely from individual-to-individual [12]. However, It can be deduced that since tumor evolution is likely to be non-linear, and substantial genetic heterogeneity is expected in tumor cell populations, this heterogeneity will be reflected epigenetically and hence may be treated in-vivo by in-vitro guidance assays. This forms the basis of individualized/personalized medicine, in which one takes the diagnostic information from a person’s own cancer to develop a highly individualized treatment for a given cancer patient, rather than relying on the challenge of empiric “one-size-fits-all” treatment modalities [10].
Since DCA had been used for years to treat rare metabolic disorders and was known to be relatively safe, [6] our data demonstrates the potential for rapid translation into clinical practice. It becomes central to develop new agents that effectively kill the cancer cells and overcome drug resistance associated with hypoxia and mitochondrial respiratory defects. Furthermore, these agents should favor cytolysis rather than cytostatic effects, so that tumor cell populations are actually killed and not merely “dazed”, if one is to achieve totally eradication of the tumor. However, if the anticancer agent is cytostatic, long-term use may still yield acceptable clinical outcomes and augmented survival rates keeping the patient in a chronic “stable” state.
Simultaneously, controlled clinical trials of DCA must be conducted to thoroughly delineate the value of DCA in cancer treatment. It is apparent that empirically-selected chemotherapy has tremendous room for improvement, since the published response rates are low in many types of cancers especially if metastaticb [39] The identification and stratification of patients to predict DCA benefit and response can easily be performed in vitro, prior to in vivo administration [40]. Toxicity is the main reason for the high failure rate (40–50%) [39] (and acquired resistance), of chemotherapeutic interventions thus, predicting how the individual oncology patient will respond to DCA (and other agents) and differentiating between direct and indirect effects [40] may be challenging but is certainly not insurmountable. Personalized treatment remains the current endeavor as improperly treated cancer takes a huge toll on our healthcare system and, more importantly, on the lives of patients and their families. Improving response rates and survival must be a priority. Thus, the initiation of new focused clinical trials containing strong correlative science components on a range of cancer patients becomes fundamental.
Abbreviations
IDC=invasive ductal carcinoma,
NSCLC= non-small cell lung cancer,
*chlor-Chlorambucil;
ix-Ixempra;
lap-Lapatinib;
lom-Lomustine;
TMZ-Temozolomide;
eto-Etoposide;
met-Metformin;
riba-Ribvirin;
rapa-Rapammune;
tam-Tamoxifen;
cis-Cisplatin;
tar-Tarceva;
MTX-Methotrexate;
dox-Doxorubicin;
tax-Taxol;
fem-Femara;
chlor-Chloroquine;
FU-Fluorouracil;
mito-Mitomycin;
vin-vinblastine;
carbo-Carboplatin;
gem-Gemcitabine;
nav-Navelbine;
iri-Irinotecan;
oxi-Oxilaplatin;
HD-High Drug Concentration = 10X Peak Plasma Concentration;
LD = Low Drug Concentration = 50% Peak Plasma Concentration
References
- Resnick MJ, Koyama T, Fan KH, Albertsen PC, Goodman M, et al. (2013) Long-term functional outcomes after treatment for localized prostate cancer. N Engl J Med 368: 436–445. [crossref]
- WARBURG O (1956) On respiratory impairment in cancer cells. Science 124: 269–270. [crossref]
- Krebs HA (1972) The Pasteur effect and the relations between respiration and fermentation. Essays Biochem 8: 1–34. [crossref]
- Gatenby RA1, Gillies RJ (2004) Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4: 891–899. [crossref]
- ED Michelakis, L Webster, J R Mackey, Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. British Journal of Cancer 99: 989–994, 2008
- Stacpoole PW, Kurtz TL, Han Z, Langaee T (2006) Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Advanced drug delivery reviews 60: 1478–87.
- Luft FC (2001) Lactic acidosis update for critical care clinicians. J Am Soc Nephrol 12 Suppl 17: S15–19. [crossref]
- Pfeffer G, Majamaa K, Turnbull DM, Thorburn D, Chinnery PF (2012) “Treatment for mitochondrial disorders”. In Chinnery, Patrick F. The Cochrane database of systematic reviews 4: 2012.
- Kato M, Li J, Chuang JL, Chuang DT (2006) Distinct Structural Mechanisms for Inhibition of Pyruvate Dehydrogenase Kinase Isoforms by AZD7545, Dichloroacetate, and Radicicol. Structure 15: 992–1004, 2006.
- Hanauske AR (2000) In: Vitro and In Vivo Predictive Tests;, Chpt 43, Holland-Frei Cancer Medicine. 5th edition, Hamilton (ON): Editor: BC Decker.
- Bonnet S, Archer SL, Michelakis ED, et al. (2007) A Mitochondria-K+ Channel Axis Is Suppressed in Cancer and Its Normalization Promotes Apoptosis and Inhibits Cancer Growth. Cancer Cell Volume 37–51, 2007
- Semenza GL (2007) Hypoxia and cancer. Cancer Metastasis Rev 26: 223–224. [crossref]
- Delgado T, Carroll, PA, Punjabi, AS, et al, (2010) Induction of the Warburg effect by Kaposi’s sarcoma herpesvirus is required for the maintenance of latently infected endothelial cells. Proc Natl Acad Sci U S A. 107: 10696–10701.
- Christofk, HR, Vander Heiden, MG, et al, (2008) The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumor growth. Nature 452: 230–233.
- PLoS One (2011) : e22445 [German] Epub 2011 Jul 26.
- Haruko Obokata, Teruhiko Wakayama, et al (2014) Stimulus-triggered fate conversion of somatic cells into pluripotency. Nature 505: 2014.
- Parra-Bonilla, G, Alvarez, DF, Cioffi, DL, Stevens, T (2008) Lung microvascular endothelial cells utilize aerobic glycolysis (e.g. Warburg effect) to sustain rapid proliferation. The FASEB Journal. 22: 1178–9.
- Morris VA, Punjabi AS, Lagunoff M (2008) Activation of Akt through gp130 receptor signaling is required for Kaposi’s sarcoma-associated herpesvirus-induced lymphatic reprogramming of endothelial cells. J Virol. 82: 8771–8779.
- Wang L, Damania B (2008) Kaposi’s sarcoma-associated herpes virus confers a survival advantage to endothelial cells. Cancer Res 68: 4640–4648.
- Sadagopan S, et al. (2007) Kaposi’s sarcoma-associated herpesvirus induces sustained NF-kappaB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression. J Virol 81: 3949–3968.
- Pfeffer G, Majamaa K, Turnbull DM, Thorburn D, Chinnery PF (2012) Treatment for mitochondrial disorders. Cochrane Database Syst Rev : CD004426. [crossref]
- Fischer, K, Hoffmann, P, Voelkl, S, et al. (2007) Inhibitory effect of tumor cell–derived lactic acid on human T cells. The dichloroacetate ion stimulates the activity of the enzyme pyruvate dehydrogenase by inhibiting the enzyme pyruvate dehydrogenase kinase. Blood 3812–3819.
- Plas DR, Thompson CB (2002) Cell metabolism in the regulation of programmed cell death. Trends Endocrinol Metab 13: 75–78. [crossref]
- Kim JW, Dang CV (2005) Multifaceted roles of glycolytic enzymes. Trends Biochem Sci 30: 142–150. [crossref]
- Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, et al. (2004) Akt stimulates aerobic glycolysis in cancer cells. Cancer Res 64: 3892–3899. [crossref]
- Pastorino L, Cusano R, Nasti S, et al. (2005) Molecular characterization of Italian nevoid basal cell carcinoma syndrome patients. Hum Mutat 25: 322–3, 2005.
- Babu, E, Ramachandran, S, CoothanKandaswamy, V, et al. (2011) Role of SLC5A8 a plasma membrane transporter and a tumor suppressor, in the antitumor activity of dichloroacetate Oncogene, 30: 4026–403.
- Georg T Wondrak (2009) Redox-Directed Cancer Therapeutics: Molecular Mechanisms and Opportunities. Antioxid Redox Signal 11: 3013–3069.
- Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Michelakis ED, et al. (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11: 37–51, 2007.
- Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I (2008) Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol 109: 394–402. [crossref]
- Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, et al. (2008) Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate 68: 1223–1231. [crossref]
- Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL (2010) Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. Int J Cancer 127: 2510–9.
- Bradford SA, Khan A (2013) Individualizing Chemotherapy using the Anti-Diabetic Drug, Metformin, as an “Adjuvant”: An Exploratory Study. J Cancer Sci Ther 5: 120–125. 2013.
- Chen LB(1988) Mitochondrial membrane potential in living cells. Annu Rev Cell Biol 4: 155–181. [crossref]
- Summerhayes IC, Lampidis TJ, Bernal SD, Nadakavukaren JJ, Nadakavukaren KK, et al. (1982) Unusual retention of rhodamine 123 by mitochondria in muscle and carcinoma cells. Proc Natl Acad Sci U S A 79: 5292–5296. [crossref]
- Sun RC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC (2010) Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat 120: 253–60. 2010.
- Yongzhuang Su (2014) Cancer Chemosensitivity Testing: Review. Journal of Cancer Therapy 5: 672–679.
- Vermorken JB, Specenier P (2010) Optimal treatment for recurrent/metastatic head and neck cancer. Ann Oncol 21 Suppl 7: vii252–261. [crossref]
- Venil N. Sumantran (2011) Cellular Chemosensitivity Assays: An Overview Methods in Molecular Biology Volume 731: 219–236.
- Verma, M, Khoury, MJ, Ioannidis, JPA (2013) Opportunities and Challenges for Selected Emerging Technologies in Cancer Epidemiology: Mitochondrial, Epigenomic, Metabolomic, and Telomerase Profiling Cancer Epidermal Biomarkers Prev 22; 189.