DOI: 10.31038/CST.2017215
Abstract
Laryngeal squamous carcinoma (LSC) represents one of the refractory malignant tumors of head and neck squamous cell carcinoma (HNSCC), leading to high patient mortality and unsatisfactory overall 5-year survival. New therapeutic targets and modalities for effective chemoprevention and treatment are required to improve the survival of patients with LSC. Evidence from previous studies has suggested that proteasome inhibitors (PIs) possess effective antitumor activity and have been approved by the United States Food and Drug Administration (FDA) to treat myeloma. However, resistance of myeloma to PIs has been reported, which prevents PIs from their clinical uses in cancer treatment. So far, the underlying mechanisms of tumor cells anti-PIs effects remain unclear. In the present study, we found that constitutive activation of signal transducer and activator of transcription 3 (STAT3) was associated with clinicopathlologic factors in LSC patients, and persistent activation of STAT3 was found in Hep-2 laryngeal carcinoma cells. Moreover, we demonstrated that proteasome inhibitor MG-132 induces apoptosis and cell cycle arrest in Hep-2 cells, accompanied by increased activation of STAT3; knockdown of STAT3 expression by shRNA or application of STAT3 inhibitor AG490 potentiates the antitumor effects of MG-132 in vitro, which is attributed to the down-regulation of cyclin D1 and Bcl-2. Taken together, we identified that STAT3 activation plays a great role in therapeutic resistance to PIs in LSC; blocking STAT3 activation could potentiate the tumor killing effects of PIs in LSC or other malignancies involving STAT3 activation. Combined application of PIs and STAT3 inhibitor may implicate a promising strategy for managing LSC or even other HNSCC.
Keywords
Signal transducer and activator of transcription 3, STAT3; proteasome inhibitor; STAT3 inhibitor; laryngeal carcinoma; apoptosis
Introduction
Laryngeal squamous carcinoma (LSC) is a kind of head and neck squamous cell carcinoma (HNSCC), the latter of which ranks the sixth most common malignant cancer, and accounts for 35,00000 newly diagnosed cases annually worldwide [1]. Despite of successive advancement in diagnosis and treatment, the overall survival of HNSCC has just been slightly improved in recent years. In addition to distant metastasis and relapse, resistance to traditional therapeutic modalities has been the dominant cause of treatment failure, especially in advanced cases. For combating the therapeutic resistance of cancers, some novel concepts and strategies for targeted therapies have been introduced, most of which mainly focused on some key modulators of cell proliferation and apoptosis pathways. Although some preliminary results have been achieved, they are not as effective as expected.
Signal transducer and activator of transcription 3 (STAT3), a member of STAT family, is a focus of many oncogenic receptor tyrosine kinase pathways, such as EGFR, IL-6/JAK and Src [2]. It has been demonstrated that STAT3 can induce up-regulation of prosurvival proteins including Bcl-2, Bcl-xL, survivin and Mcl-1, and promote expression of angiogenesis factors, such as VEGF [3]. In fact, STAT3 is constitutively activated in many tumors [4-7] and plays a great role in proliferation, cell cycle progression and apoptosis of tumor cells [8]. In addition, activation of STAT3 is also associated with chemoresistance in many malignancies [9, 10]. Results from our previous study demonstrate that knockdown of STAT3 with specific shRNA can enhance radiosensitivity in LSC Hep-2 cells both in vitro and in vivo [11, 12]. Therefore, targeting STAT3 is a potential therapeutic strategy for LSC.
As is known, due to rapid proliferation and insufficient blood supplies, the microenvironments of most solid cancers are accompanied by hypoxia, glucose starvation and low PH, leading to endoplasmic reticulum (ER) stress due to causal accumulations of incorrect proteins [13]. Sequently, cancer cells must adapt to these unfavorable conditions for their survival via inhibiting synthesis of proteins and degrading unfolded proteins accumulated in ER by proteasome [14]. Therefore, targeting proteasome becomes a promising strategy for cancer therapy.
Early studies demonstrated that tumor-inhibiting effects of proteasome inhibitors (PIs) are attributed to repression of nuclear factor-κB (NF-κB) signaling and regulation of proteins associated with both cell cycle and apoptosis, such as p27Kip1 [15], p21cip/WAF1 [16], p53 [17], Bcl-2 [18, 19], Bax [17], Bim and Bik [20]. It is now believed that PIs induce ER stress-related cell death through unfolded protein response (UPR) [21] and confer preferential cytotoxicity towards hypoxic tumor cells [22]. However, cases of resistance to PIs are emerging, whereas the related mechanisms are not systematically elucidated [23]. Interestingly, a recent study has demonstrated that a proteasome inhibitor Bortezomib promotes STAT3 activation in HNSCC, suggestive of potential roles of STAT3 in the resistance of HNSCC to the proteasome inhibitor [24]. Therefore, it would be of great interest to elucidate whether the STAT3 plays a role in resistance LSC to PIs.
In the present study, we first assessed the associations between constitutive activation of STAT3 and clinicopathlological factors in LSC. We next investigated the role of STAT3 activation by protesome inhibitor and its impacts on proteasome-induced cell apoptosis in LSC.
Materials and Methods
Patients’ data
50 LSC patients (36 males and 14 females) who received surgical resection at Bethune International Peace Hospital were recruited, and their complete detailed clinicopathologic data were collected. Of the 50 cases, there were 17 cases of supraglottic and 34 cases of glottic cancers. The age of the present series ranged from 36 to 86 years, with a mean age of 58 years. All patients had undergone neither chemotherapy nor radiotherapy prior to surgery. According to UICC cancer staging system (2002), there were 7 cases of stage I, 6 cases of stage II, 17 cases of stage III and 20 cases of stage IV tumors. With regard to the histological grading, 6 cases were well differentiated, 13 cases were moderately differentiated and 31 cases were poorly differentiated squamous cell carcinomas (SCC). Twenty-two cases had neck node metastasis as proven by postoperative pathologic examinations.
Tumor tissue collection
With the informed consent of all patients, tissue specimens were collected immediately after surgical removal. Paired tumor-free paracancerous tissues (PCT) were taken at least 0.5 cm beyond the tumor margins. Each sample was cut in halves. One half was promptly frozen at -70 ℃ for subsequent Western blot, while the other was fixed immediately with 10% neutrally buffered formalin for histopathologic evaluation and immunohistochemistry. The acquisition of paraffin tissues and frozen specimens were approved by the Institutional Review Boards at Bethune International Peace Hospital. Neck dissection specimens were evaluated by two pathologists to confirm the status of neck node metastasis in these patients.
Immunohistochemistry and immunocytochemistry
Immunohistochemical and immunohistocytochemical studies were performed as described previously [25] using SP-9002 kit (including 0.3% hydrogen peroxide, 10% goat serum, biotinated secondary antibodies and horseradish peroxidase marked strepto- antibiotin, Zhong Shan Jin Qiao, Beijing) with some modifications. Briefly, for immunohistochemistry, consecutive sections were deparaffinized and hydrated, and subjected to 0.3% hydrogen peroxide for elimination of endogenous peroxidase at room temperature for 10 min. Slides were then heated for the retrieval of antigens in an autoclave sterilizer for 2 min. Goat serum (10%) was added to block nonspecific protein staining for 10 min. The sections were incubated with anti-STAT3 and anti-p-STAT3 (Santa Cruz Biotechnology Inc.) at 4 ℃overnight. Afterwards, they were rinsed by PBS and incubated at room temperature with biotinated secondary antibodies for 15 min under a humid condition followed by 15-min incubation with horseradish peroxidase-marked strepto- antibiotin at room temperature. Finally, they were detected with DAB kit (Zhong Shan Jin Qiao, Beijing). The immunoreactivity of STAT3 or p-STAT3 was evaluated as described previously [31]. Brown coloration was identified as positive staining. The distribution of positive staining cells was classified in 4 categories: weakly positive (+), positive (++), strongly positive (+++), and negative. For immunocytochemical study, Hep-2 cells were seeded in 6-well plates with a small cover slide in each well. When confluence reached 70% to 80%, cells grown on the cover slide were fixed with cold acetone at room temperature for 15 min. Cover slides were then subjected to 0.3% hydrogen peroxide for elimination of endogenous peroxidase at room temperature for 30 min, washed with PBS and permeabilized with 0.1% TritonX-100 on ice for 2min. The immunostaining of Hep-2 cells was performed as in immunohistocehmistry.
Cell line and culture
Hep-2 human LSC cell line (National Cancer Institute, USA) were cultured in RPMI1640 medium supplemented with 10% fetal bovine serum (FBS) under the condition of 37℃, 20% O2, 5%CO2, 95% humidity in a CO2 incubator. Cells were allowed for attachment overnight and those grown in logarithmic phase were used for subsequent experiments. All experimental groups came from the same parental cells. Cells were passaged every 2 to 3 days to maintain exponential growth.
Transfection of plasmids
The construction of STAT3-targeting shRNA expression vector and the transfection of plasmids were performed as our previous publications [12]. Briefly, the double strands of shRNA targeting gene of STAT3 (sense: 5′-CACCGCAGCAGCTGAACAACATGTTCAAGAGACATGTTGTTCAGCTGCTGCTTTTTTG-3′, the corresponding mRNA coding region is underlined) and the double strands of negative control gene (sense: 5′-CACCGTTCTCCGAACGTGTCACGTCAAGAGATTACGTGACACGTTCGGAGAATTTTTTG-3′, underline shows the target sequence) were inserted into pGPU6/GFP/Neo vector (Gene Pharma Co., Ltd), and named pGPU6/GFP/Neo-shSTAT3 and pGPU6/GFP/Neo-shNC, respectively. The procedure of transient transfection was performed according to LipofectamineTM2000 (Invitrogen, Corporation) manufacturer’s instructions in 96-well plates or/and 6-well plates. The thansfection efficiency was monitored by observation under a fluorescent microscope and by flow cytometry. Sequent assays were performed 24 hours after transfection.
MTT assay
The inhibition of hep-2 cells by chemicals was determined by MTT assay as described previously [26]. Transfected and untransfected hep-2 cells were subjected to MTT assay. After treatments with PIs (MG-132) (MERCK Corporation, Germany) and STAT3 inhibitor (AG940) (Sigma, USA) in different combinations and cisplatin (DDP, Shan Dong Qi Lu Pharmaceutics, China), 20μl 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT, 5mg/ml, Sigma, USA) were added into each well of 96-well plates. After incubation at 37 ℃ for 4h, culture medium was replaced by DMSO to dissolve formazan for coloration. Absorbance was measured at 490 nm using Enzyme-linked Immunosorbent Detector (Model 550, Bio-Rad, USA).
Flow cytometry
The transfection efficiency, apoptosis rate and cell cycle distributions were determined by a FACScan analyzer (Becton Dickinson FAC Sort, USA). Hep-2 cells, with or without treatments, were collected after trypsinization. Fresh cells were used for detection of the efficacy of transfection by flow cytometry. To determine apoptosis rate and cell cycle distribution, cells were washed by PBS for 3 times, and then were fixed with 75% ethanol overnight at 4 ℃. Prior to analysis, ethanol was discarded and 80μl RNase (final concentration: 50mg/L) and 150μl propidium iodide (final concentration: 50mg/L, Sigmar, USA) were used for staining of DNA for 30 min at 4℃. Apoptotic cells appeared as a magnitude of the sub-G1 peak due to nuclear fragmentation and loss of DNA.
Western blot
Western blot was carried out as described previously [25] with minor modifications. For tissue specimens, lysates were obtained from 100mg sample of each frozen tissue by a homogenizer. Subjected Hep-2 cells were harvested following trypsinization and centrifugalization. The cell pellets were resuspended in cell lysis buffer containing 500mM HEPES 25ml (0.1mol/L),100mM NaCl 5ml (1mol/L),2mM EDTA 0.2 ml (0.5mol/ L),sodium deoxycholate 0.25g (0.5%),NP-400 5ml (1%),10%SDS 0.5ml (0.1%),2×10-3 mol/L PMSF and 2×10-6 g/L Lupeptin to collect lysates. Subsequently, lysates originating from tissues or hep-2 cells were acquired in the same way. After determination of protein concentrations in the lysates, equivalent protein (100mg from tissues or 30mg from cells) were separated by electrophoresis on 4% to 10% SDS-PAGE gels. The subjected gels were then transferred onto nitrocellulose filters, and blocked in confining liquid containing 1% nonfat dry milk, 0.02% Tween-20 (Sigmar, USA), 10mmol/LTris-Cl,0.15mmol/L NaCl at 4℃ overnight. The blocked filters were incubated with primary antibodies (Santa Cruz Biotechnology int and Cell Signaling Technology, USA) at 4℃ overnight followed by rinse in TBST. The filters were incubated with secondary antibodies marked by HRE (Zhong Shan Jin Qiao, BeiJing) at room temperature for 1 hour followed by rinse in TBST two times. Finally, the blots were visualized by ECL kit (Pierce, USA).
Statistical analysis
Results were analyzed for statistical significance with the use of SPSS 13.0. Data were presented as mean values ± SD. The method of statistical analysis included student’s t-test for comparision of two groups, one-way ANOVA for comparison of more than two groups combined with or without least significant difference (LSD) followed for comparison of each two groups, and Pearson correlation analysis for correlation evaluation of different factors. P < 0.05 was considered significant.
Results
Constitutive activation of STAT3 is associated with clinicopathlological factors in LSC, including clinical stage, pathological grading and cervical lymph node metastasis. It has been reported that STAT3 is aberrantly activated in many tumors in comparison with its transient activation in normal tissues [27-30]. To determine activation status and expression distribution pattern of p-STAT3 in LSC, we compared levels of p-STAT3 protein of primary LSC tissues with corresponding tumor-free PCT by immunohistochemistry. As can be seen in Fig 1A-D, p-STAT3 was constitutively expressed in LSC compared with PCT. In positive samples, weakly positive staining dominates PCTs, whereas strong positive coloration occupies most of cancer tissues; it was also noted that the staining of p-STAT3 in poorly differentiated LSC is stronger than that in well differentiated LSC.
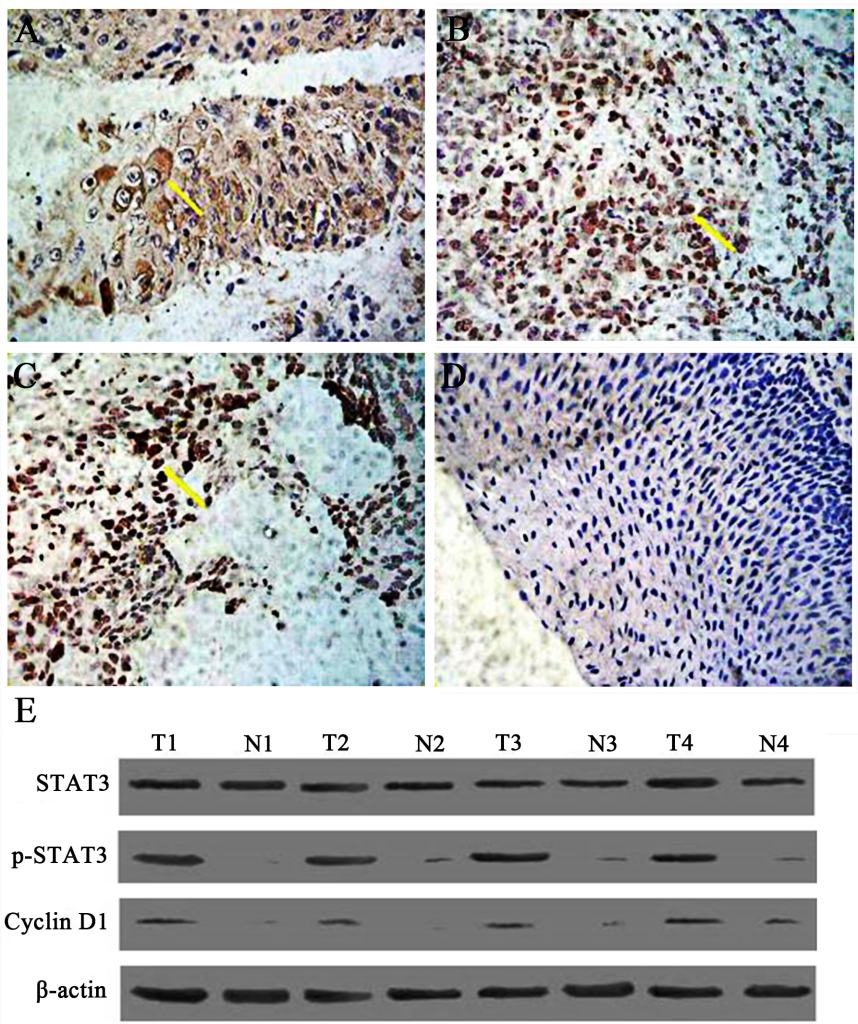
Figure 1. The expression of phosphorylated STAT3 (p-STAT3) in human laryngeal squamous cell carcinoma (SP×400). Tissue specimens were collected and p-STAT3 protein expression levels were assessed by immunohistochemistry and western blot. A. Expression of STAT3 in well-differentiated laryngeal squamous carcinoma. B. The expression of p-STAT3 protein in moderately-defferentiated laryngeal squamous cell carcinoma (SP×400). C. The expression of p-STAT3 protein in poorly differentiated laryngeal squamous cell carcinoma (SP×400). D. The expression of p-STAT3 protein in tumor-free paracancerous laryngeal epithelial tissues (SP×200). Yellow arrow indicates representative staining cells. E. Expression of Stat3, p-STAT3, and cyclin Dl in human laryngeal carcinoma by western blot. Elevated levels of p-STAT3 and cyclin Dl in tumor tissues (T) compared to adjacent normal laryngeal mucosa (N) were further confirmed.
These findings suggest that STAT3 is activated constitutively and persistently in LSC rather than in PCT, and its activated form, p-STAT3, is likely to function in nucleus as in other malignancies. Moreover, expression of p-STAT3 is closely related to the expression of cyclin D1. Results from western blot analysis showed the same expression pattern for each protein (Fig 1E) in that there is a correlation between p-STAT3 and the expression of cyclin D1, suggesting that cyclin D1 is regulated by p-STAT3 in LSC tissues.
It has been reported in many studies that activation of STAT3 is related to poor prognosis of various cancers [31-33]. To test whether STAT3 activation is pertinent to clinicopathologic factors of LSC, we analyzed the relationship between p-STAT3 and critical prognostic factors. Results in Table 1 indicates that there is a significant correlation between p-STAT3 expression and clinical stage, pathologic grade and neck node metastasis rather than age, sex, T stage and even the site of primary tumors.
Table 1. The relationship between the expression of Stat3 and phosphorylated Stat3 protein and clinicophalogical features in laryngeal squamous cell carcinoma
| n | Stat3 | p-Stat3 | ||||
| M number (rate%) | P value | M number (rate%) | P value | |||
| Age | ≥60 | 28 | 23(82.14) | 0.0715 | 17(60.71) | 0.6609 |
| <60 | 22 | 13(59.09) | 12(54.54) | |||
| Sex | M | 36 | 27(75.00) | 0.6841 | 23(63.89) | 0.1761 |
| F | 14 | 9 (64.29) | 6 (42.86) | |||
| Tumor position | Supra-glottis | 17 | 10(58.82) | 0.2473 | 10(58.82) | 0.9325 |
| glottis | 33 | 26(78.78) | 19(57.58) | |||
|
Patho- grade |
Well to moderately differentiated | 19 | 10(52.63) | 0.0169 | 5 (26.32) | 0.0004 |
| Poorly differentiated | 31 | 26(83.87) | 24(77.42) | |||
| T stage | T1-T2 | 17 | 12(70.58) | 0.0685 | 7 (41.18) | 0.0836 |
| T3-T4 | 33 | 27(81.81) | 22(66.67) | |||
| Clinical stage | Ⅰ-Ⅱ | 13 | 5 (38.46) | 0.0056 | 4 (30.77) | 0.0208 |
| Ⅲ-Ⅳ | 37 | 31(83.78) | 25(67.57) | |||
| Lymph node metastasis | Yes | 22 | 20(90.90) | 0.0083 | 17(77.27) | 0.0144 |
| No | 28 | 16(57.14) | 12(42.86) | |||
Activation of STAT3 is in constant existence in Hep-2 laryngeal carcinoma cells (Hep-2 cells). To test whether STATA3 is also constitutively activated in Hep-2 cells, we examined the expression of STAT3 and its related proteins by immunocytochemistry and western blot. By immunocytochemistry, STAT3 was distributed in cytoplasm of cancer cells, while p-STAT3 was observed mostly within nucleus of Hep-2 cells, which resembles the expression pattern of STAT3 and p-STAT3 in LSC tissues (Fig 2A, B, C). To examine if these proteins were constantly and pervasively expressed, we detected expression of the two proteins using western blot at different time points after the setting of cell culture. The sequent 24 h, 48 h, and 72 h were chosen as time spots of analysis. The results of western blot showed that STAT3 and p-STAT3 (Fig 2D) can be detected at different time points, suggesting that constitutive activation of STAT3 is also present in Hep-2 cells as in primary LSC.
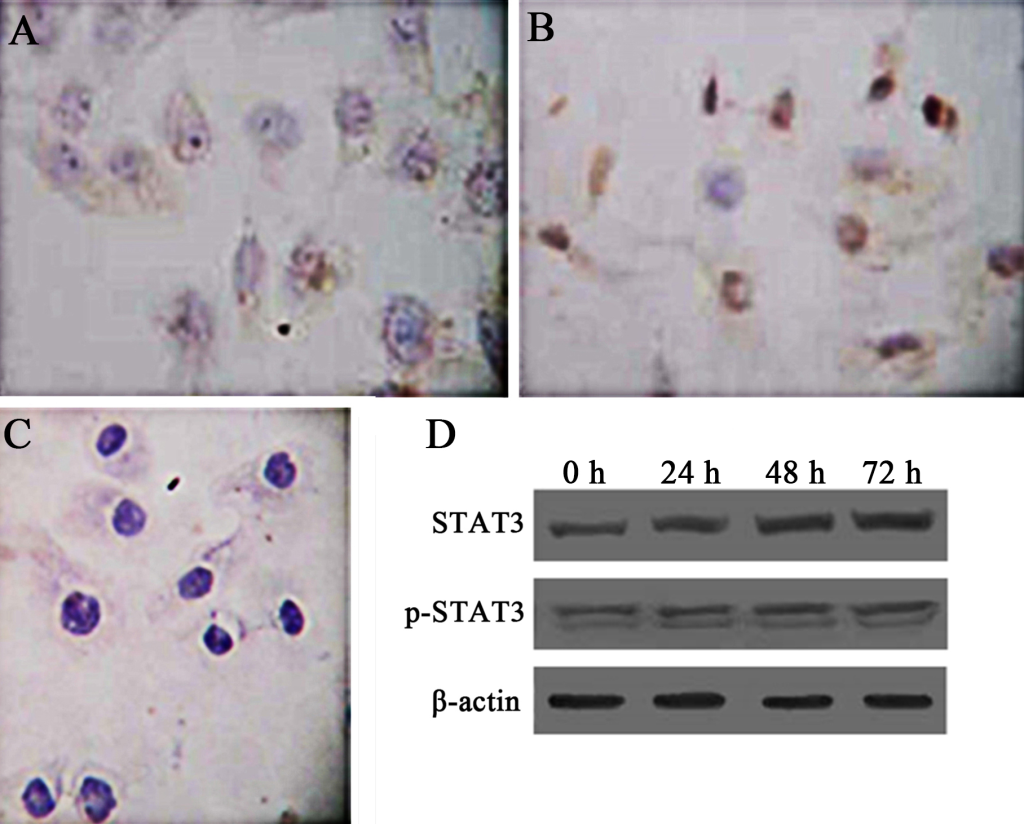
Figure 2. TExpression of STAT3 and p-STAT3 in human laryngeal carcinoma Hep-2 cells. Immunocytochemistry was used to demonstrate the intracellular distribution of STAT3 and p-STAT. PBS was used as a negative control. Besides, western blot was used to show the expression patterns of STAT3 and p-STAT3 overtimes (0h, 24h, 48h, 72h). β-actin represents the internal protein control. Constitutive expression and activation of STAT3 in human laryngeal carcinoma Hep-2 cells was noted. A. Cytoplasmic staining of STAT3 in Hep-2 cells (SP × 400). B. Nuclear staining of p-STAT3 in Hep-2 cells (SP × 400). C. Negative control (SP × 400). D. Expression of Stat3 and p-STAT3 in Hep-2 cells as demonstrated by western blot.
Proteasome inhibitor MG-132 induces apoptosis in Hep-2 cells, which is accompanied by additional activation of STAT3 and changes in the expression of several proteins related to cell cycle and apoptosis regulation. PIs have been approved by the United States Food and Drug Administration (FDA) for the treatments of multiple myeloma. In addition, PIs have been investigated for the treatments of many cancers. Herein, we aimed to test the therapeutic effects of PIs on Hep-2 cells. First, we tested the inhibition of proliferation in Hep-2 cells by a PI, MG-132, using MTT assay. Medium containing different concentrations of MG-132 was add into each well of treatment groups and maintained for different times. As seen in Fig 3A, the inhibition of Hep-2 cells by MG-132 displayed a concentration-dependent manner (24h:r=0.925,p<0.01;48h:r=0.944,p<0.01). Second, we attempted to confirm if MG-132 inhibits proliferation of Hep-2 cells in a time-dependent manner. For doing this, we selected the 2.5μM (IC50) of MG-132 to stimulate Hep-2 cells for 48 h. The results of Fig 3B show that the effects of MG-132 in proliferation inhibition is enhanced with prolongation of treatment time (r=0.945,p<0.01). Third, using flow cytometry, we determined apoptosis rate and cycle distribution of Hep-2 cells after treatment with MG-132. Similar with the inhibition of proliferation, MG-132 induces apoptosis in time- (r=0.888,p<0.01) and dose-dependent (r=0.842,p<0.01) manners (Fig 3C, 3D ).
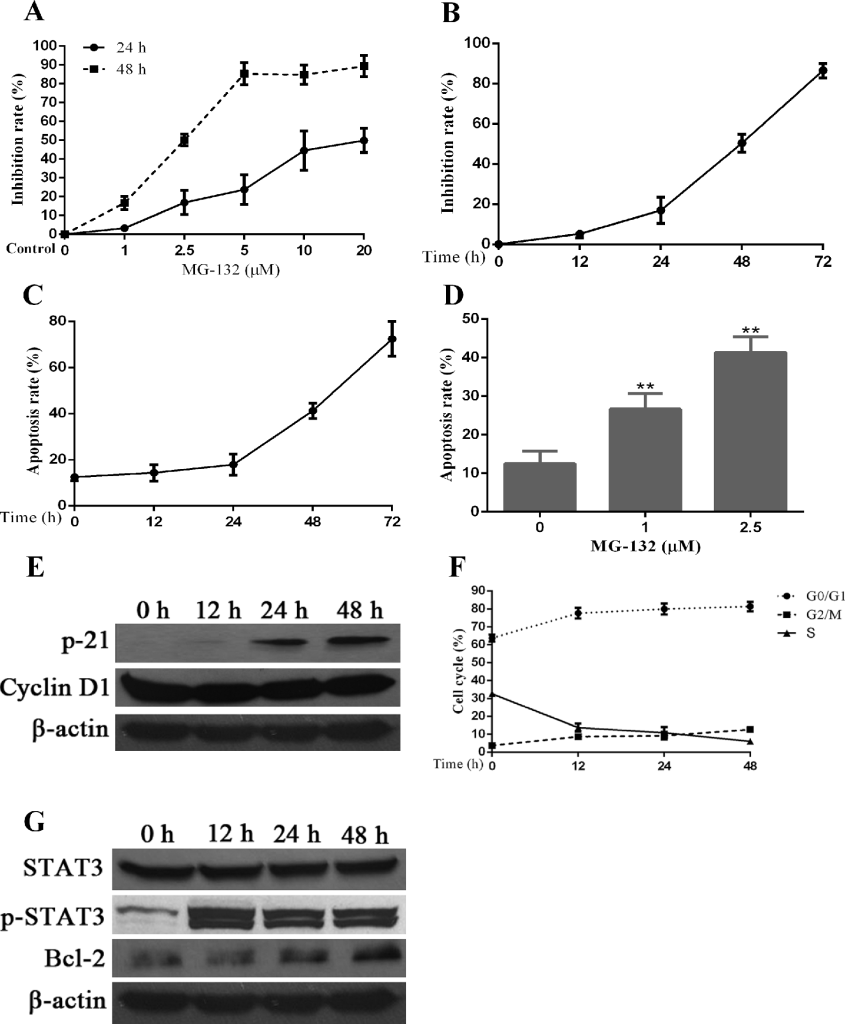
Figure 3. Effects of a proteasome inhibitor, MG-132, on Hep-2 cells. Hep-2 cells were treated with different concentrations of MG-132 for 24 hours. A concentration of 2.5μM was chosen for stimulation of Hep-2 cells for different times. Alterations in cell apoptosis, cell cycle and protein expression of STAT3 and p-STAT3 were assessed simultaneously. A. The dosage and effect curve of MG-132 (n = 3). B. The time and effect curve of MG-132 (n = 3). C. Apoptosis rate of Hep-2 cells treated with MG-132 for different time (n = 3). D. Apoptosis rate of Hep-2 cells treated with MG-132 at different concentrations (*P < 0.05, versus untreated groups (0μM) (n = 3). E. The correlated expression of cell cycle regulation proteins in Hep-2 cells treated with MG-132 for different time. F. Variation of cell cycle of Hep-2 cells treated with MG-132 for different time (n = 3). G. MG-132-induced STAT3 activation and Bcl-2 expression in Hep-2 cells.
To confirm the mechanisms underlying the cell cycle changes induced by MG-132, we used western blot to check the expression levels of some cell cycle regulators. As seen in Fig 3E, the expression of P21 is elevated significantly compared with the decreased levels of the expression of cyclinD1, suggesting that the changes of cell cycle induced by MG-132 were associated with upregulation of P21 and downregulation of cyclinD1. Simultaneously, cell cycle arrest was noted at G0/G1 phase and/or G2/M phase overtimes after treatment with MG-132 (Fig 3F), the former of which is more prominent than the latter.
From the results of western blot in Fig 3G, we can confirm that STAT3 and its activated form, p-STAT3, are expressed at all times in cultured Hep-2 cells, even in the absence of MG-132 treatment, and the level of its expression was relatively constant and stable. In the presence of MG-132, the expression of p-STAT3 was elevated within short period of time and remained at a stable level; however, the increased expression of p-STAT3 was not related to the duration of MG-132 treatment. Bcl-2 was identified as a downstream of STAT3, and facilitates anti-apoptosis activity in cancer cells [34]. Expression of Bcl-2 protein increased overtime after MG-132 treatment, which may contributed to the MG-132-induced STAT3 activation and resistance of Hep-2 cells to MG-132-induced apoptosis.
Knockdown of STAT3 by shRNA, abrogates the expression and thus activation of STAT3, and potentiates the apoptosis-inducing effects of MG-132. To further confirm the role of the STAT3 activation in inducing resistance of Hep-2 cells to MG-132, we used RNAi to knockout the expression of STAT3, and thus the “constitutive” (inherent) and “PI-induced” (acquired) activation of STAT3. The transfection efficiency was 85.76% by flow cytometry (data not shown). The results of Fig 4A showed that combined group has a similar expression level of p-STAT3 with pshSTAT3 group, with a higher inhibition rate than any other groups (all p<0.01). Results from apoptosis detection Fig 4B showed a more striking apoptosis rate 55.80±3.12% in combined group than in any other group (all p<0.01). In pshSTAT3 group, moderate apoptosis and proliferation inhibition rates were induced. Therefore, blockade of STAT3 by shRNA, abrogates the expression and thus both “inherent” and “acquired” activation of STAT3, and potentiates the MG-132-induced apoptosis in Hep-2 cells.
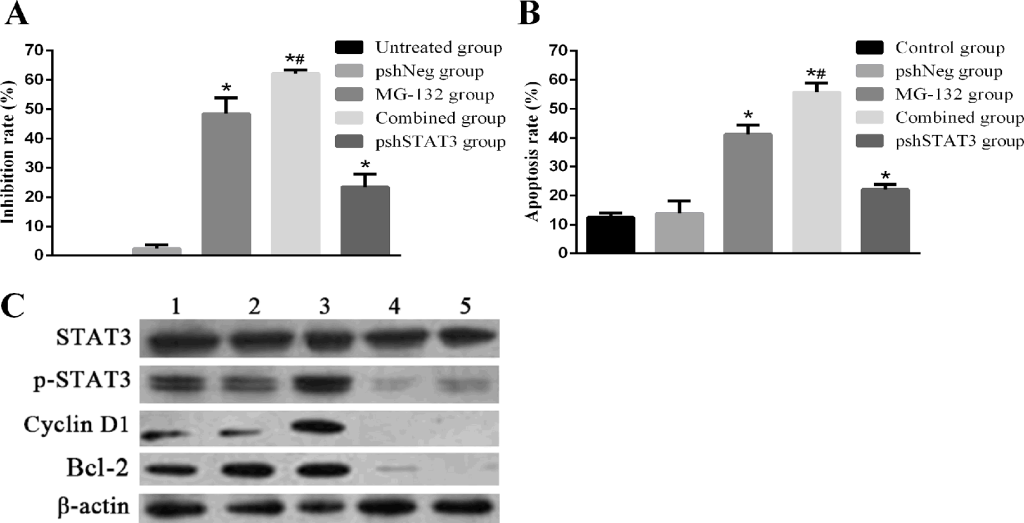
Figure 4. Effects of MG-132 combined with STAT3 RNAi on proliferation inhibition and apoptosis in Hep-2 cells, as assessed by MTT assay and flow cytometry. Hep-2 cells were seeded into 96-well plates for MTT assay, or 6-well plates for flow cytometry. Subjected Hep-2 cells were categorized as five groups: normal control group (untreated Hep-2 cells), negative control group (Hep-2 cells transfected with negative plasmid, pshNeg group), MG-132 group (Hep-2 cells treated with 2.5μM MG-132 only), combined group (Hep-2 cells transfected with positive plasmid combined with 2.5μM MG-132 treatment), pshSTAT3 group (Hep-2 cells transfected with positive plasmid without any other treatment). A. The proliferation inhibition rate of Hep-2 cells in different groups (n=3) (versus pshNeg group, all *P<0.01; pshSTAT3 group versus MG-132 group, #P<0.01). B. The apoptosis rate of Hep-2 cells in different groups (n=3) (control group versus pshNeg group, *P<0.01; pshSTAT3 group versus MG-132 group, #P<0.01). C. Expression of p-STAT3, cyclin D1 and Bcl-2 in Hep-2 cells in different groups was analyzed by Western blotting.. Lane 1: control group; lane 2: pshNeg group; lane 3: MG-132 group; lane 4: combined group; lane 5: pshSTAT3 group.
With regard to the activation of STAT3 at different situations, as seen in Fig 4C, p-STAT3 was expressed at the highest level in MG-132 group. By contrast, p-STAT3, cyclin D1, and Bcl-2 were expressed at lower levels in combined group, compared with each of the normal control group, negative control group and MG-132 group. The similar expression and activation patterns of STAT3 noticed in combined group and pshSTAT3 group indicated that p-STAT3 may participate in the formation of resistance of Hep-2 cells to MG-132 at a transcriptional level. Taken together, activation of STAT3 is responsible for the resistance of Hep-2 cells to MG-132 at least in part by regulating the anti-apoptosis factor Bcl-2 expression.
Combined treatment with STAT3 inhibitor AG490 and proteasome inhibitor MG-132 induces apoptosis more effectively in Hep-2 cells. In view of the fact that activation of STAT3 played a role in the therapeutic resistance to MG-132 in Hep-2 cells, which can be sensitized by knockdown of STAT3, we studied the effects of combined using STAT3 inhibitor AG490 and proteasome inhibitor MG-132 on proliferation and apoptosis. As seen in Fig 5 A, B, combination treatment by AG490 and MG-132 can induce significant increase in cell proliferation inhibition and apoptosis, indicative of effective synergism.
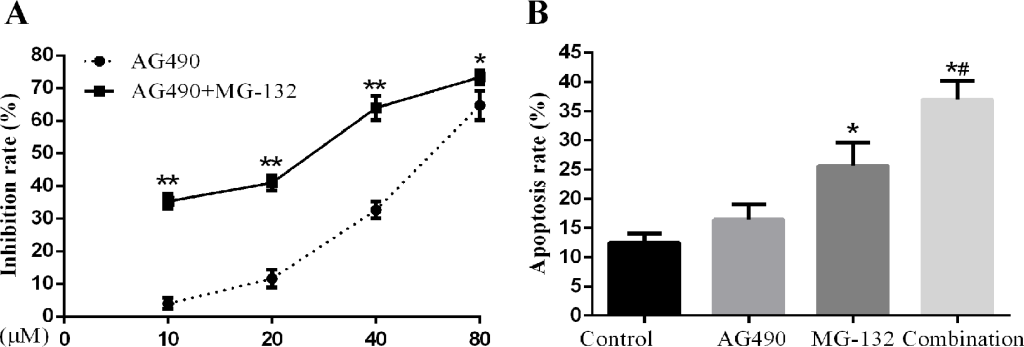
Figure 5. Effects of AG-490 combined with MG-132 on Hep-2 cells. Hep-2 cells were seeded into 96-well or 6-well plates followed by treatment with 1μM of MG-132 for 48h with or without presence of different concentrations (10, 20, 40, 80μM) of AG490. A concentration of 10μmol/L was for analysis of AG490-induced apoptosis. A. The proliferation inhibition rate of Hep-2 cells treated with the different concentrations AG490 alone and in combination with MG-132 for 48h (n=3). B. The apoptosis rate of Hep-2 cells treated with AG490 alone and in combination with MG-132(1μM) for 48h (n=3) (compared with control group,*P<0.01; compared with AG490 group and MG-132 group, all #P<0.01).
Discussion
Like other malignancies in HNSCC, LSC often exhibits resistance to various therapeutic strategies including chemoradiation and targeted therapies, especially in advanced cases. This difficult situation is often attributed to aberrant expression and activation of some specific genes, such as STAT3, which are utilized by cancer cells to maintain their biological behaviors. In this regard, cancer cells may become vulnerable to the assigned treatment modalities secondary to abrogating the functions of major responsible oncogenes, the so-called “Achilles heel”, which constitutes the fundamental rationale for targeted therapy [35].
Previous studies have demonstrated that activation of STAT3 is associated with chemo- and/or radioresistance in Hep-2 cells, and knockdown of STAT3 by siRNA or blockade of STAT3 activation by its inhibitors sensitizes chemo- and/or radiotherapy both in vitro or in vivo [11, 12, 36, 37]. Importantly, we also demonstrated in the present investigation that constitutive activation of STAT3 was associated with clinicopathlologic factors in LSC, which include clinical stage, pathological grading and cervical lymph node metastasis. This is consistent with the findings in the study of Rosen et al, in which they demonstrated that p-STAT3 was associated with poor prognosis of ovarian cancer [38]. Moreover, results of Kusaba’s study [33] also demonstrated that p-STAT3 is correlated with invasion of vein, nodal metastasis and Dukes grades instead of pathological grading in ovarian cancer. In the present study, we found expression of cyclin D1, one of the important factors downstream of STAT3, was also elevated. Taken together, correlations between activation of STAT3 and clinicopathlological factors of different cancers may depend on cell types from which they originate.
In addition to intrinsic elements, cancer cells in solid tumors also depends on extrinsic factors that form surrounding microenvironment (also called “niche”) for their proliferation and survival. The feature of the niche is characterized by low PH, hypoxia and glucose-deprived starvation as a result of rapid proliferation by tumor cells and insufficient delivery by intra-tumor capillary [39]. Under the stressful conditions, a mass of misfolded or unfolded proteins accumulate in endocytoplasmic reticulum (ER). Tumor cells must activate UPR to inhibit synthesis of protein and mRNA and degrade excessive protein by proteasome to accommodate ER-stress, which is rarely experienced by normal tissue cells [40]. It has been demonstrated that severe ER-stress caused by accumulated misfolded proteins can activate programmed death in tumor cells [39]. Therefore, proteasome can serve as a potential target for cancer therapy.
Recently, PIs was introduced into clinical treatment of cancers, either as a single agent or a combined therapy to sensitize traditional chemo- and/or radiotherapy. However, the resistance to PIs remains a problem to solve, of which underlying mechanisms have not been explained pertinently [23]. The prerequisite for us to select PIs as an anti-LSC agent resides on the fact that PIs is effective against cyclin D1 highly-expressed cancers [41]. However, it remains to be elucidated whether other HNSCC cells were dependent or independent on cyclin D1 expression for their proliferation, survival and progression.
Given the fact that activation of STAT3 is associated with clinicopathologic behaviors and chemoradiation resistance in LSC, STAT3 is definitely a potential target for therapeutic purposes. The pro-survival and –proliferation effects of p-STAT3 let us to postulate that the constitutive (inherent) along with PI-induced (acquired) activation of STAT3 may constitute a major cause of resistance to PIs in LSC. Abrogation of both existed and acquired STAT3 activation may sensitize the LSC cells to PI treatment. In present investigation, we found for the first time that activation of STAT3 contributed to resistance of LSC to MG-132 at least in part by upregulating expression of antiapoptotic protein Bcl-2, which is partly consistent with results of a recent study [24]. Our results showed that PIs plus AG490 displayed a more powerful killing effect to LSC cells in a synergetic manner. However, such a combination may not be applied to all cancers, because overexpression of Bcl-2 is associated with a good prognosis in some other malignancies [42].
In the present study, we also demonstrated that MG-132 can induce G0/G1 cell cycle arrest, via downregulation of cyclin D1 in LSC, which is also observed in some other cancers [43-45]. It can be inferred that an ideal local control is to be achieved by PIs through inducing quiescence in LSC cells. However, a fact that cannot be ignored is that G0/G1 cycle arrest also constitutes a cause of therapeutic resistance in some tumors.
Although inhibiting STAT3 is capable of inducing quiescence and potentiating PI-induced apoptosis in LSC cells, there has been no effective STAT3 inhibitor that can be safely used for humans in clinic for treatment of HNSCC. We recently defined dihydroartemisinin as putative STAT3 inhibitor, which might be safely used in clinic for cancer treatment [46]. Much work has to be done for developing new STAT3-inhibiting agents and exploring new treatment strategies and modalities based on the synergized apoptosis-inducing effects of PIs with STAT3 inhibitors.
Acknowledgements: The research work in the present investigation is supported by Key Basic Research Project Fund of Hebei Province, China (Fund No. 14967721D) and in part by Scientific Research Fund of Bethune International Peace Hospital, China.
Conflict of Interest: None
References
- Parkin DM, Bray F, Ferlay J, Pisani P (2005) Global cancer statistics, 2002. CA Cancer J Clin 55: 74-108. [crossref]
- Leeman RJ, Lui VW, Grandis JR (2006) STAT3 as a therapeutic target in head and neck cancer. Expert Opin Biol Ther 6: 231-241. [crossref]
- Chen SH, Murphy DA, Lassoued W, Thurston G, Feldman MD, et al. (2008) Activated STAT3 is a mediator and biomarker of VEGF endothelial activation. Cancer Biol Ther 7: 1994-2003. [crossref]
- Grandis JR, Drenning SD, Chakraborty A, Zhou MY, Zeng Q, et al. (1998) Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor- mediated cell growth In vitro. J Clin Invest 102: 1385-1392. [crossref]
- Guo C, Yang G, Khun K, Kong X, Levy D, et al. (2009) Activation of Stat3 in renal tumors. Am J Transl Res 1: 283-290. [crossref]
- Kim SM, Kwon OJ, Hong YK, Kim JH, Solca F, et al. (2012) Activation of IL-6R/JAK1/STAT3 signaling induces de novo resistance to irreversible EGFR inhibitors in non-small cell lung cancer with T790M resistance mutation. Mol Cancer Ther 11: 2254-2264. [crossref]
- Sánchez A, Nagy P, Thorgeirsson SS (2003) STAT-3 activity in chemically-induced hepatocellular carcinoma. Eur J Cancer 39: 2093-2098. [crossref]
- Turkson J, Jove R (2000) STAT proteins: novel molecular targets for cancer drug discovery. Oncogene 19: 6613-6626. [crossref]
- Bonner JA, Trummell HQ, Willey CD, Plants BA, Raisch KP (2009) Inhibition of STAT-3 results in radiosensitization of human squamous cell carcinoma. Radiother Oncol 92: 339-344. [crossref]
- Ara T, Nakata R, Sheard MA, Shimada H, Buettner R, et al. (2013) Critical role of STAT3 in IL-6-mediated drug resistance in human neuroblastoma. Cancer Res 73: 3852-3864. [crossref]
- Li X, Wang H, Lu X and Di B (2010) Silencing STAT3 with short hairpin RNA enhances radiosensitivity of human laryngeal squamous cell carcinoma xenografts in vivo. Exp Ther Med 1: 947-953.
- Li X, Wang H, Lu X and Di B (2010) STAT3 blockade with shRNA enhances radiosensitivity in Hep-2 human laryngeal squamous carcinoma cells. Oncol Rep 23: 345-353.
- Määttänen P, Gehring K, Bergeron JJ, Thomas DY (2010) Protein quality control in the ER: the recognition of misfolded proteins. Semin Cell Dev Biol 21: 500-511. [crossref]
- Orlowski RZ (2005) The ubiquitin proteasome pathway from bench to bedside. Hematology / the Education Program of the American Society of Hematology. American Society of Hematology. Education Program: 220-225
- Uddin S, Ahmed M, Bavi P, et al. (2008) Bortezomib (Velcade) induces p27Kip1 expression through S-phase kinase protein 2 degradation in colorectal cancer. Cancer Res 68: 3379-3388.
- Mitsiades CS, McMillin D, Kotoula V, et al (2006) Antitumor effects of the proteasome inhibitor bortezomib in medullary and anaplastic thyroid carcinoma cells in vitro. The Journal of clinical endocrinology and metabolism 91: 4013-402.
- Jones RJ, Chen Q, Voorhees PM, Young KH, Bruey-Sedano N, et al. (2008) Inhibition of the p53 E3 ligase HDM-2 induces apoptosis and DNA damage–independent p53 phosphorylation in mantle cell lymphoma. Clin Cancer Res 14: 5416-5425. [crossref]
- Li B and Dou QP (2000) Bax degradation by the ubiquitin/proteasome-dependent pathway: involvement in tumor survival and progression. Proc Natl Acad Sci U S A 97: 3850-3855.
- Lara PN, Bold RJ, Mack PC, Davies AM, Gumerlock PH and Gandara DR (2005) Proteasome inhibition in small-cell lung cancer: preclinical rationale and clinical applications. Clinical lung cancer 7 Suppl 2: S67-7
- Li C, Li R, Grandis JR and Johnson DE (2008) Bortezomib induces apoptosis via Bim and Bik up-regulation and synergizes with cisplatin in the killing of head and neck squamous cell carcinoma cells. Mol Cancer Ther 7: 1647-1655
- McConkey DJ, Zhu K (2008) Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist Updat 11: 164-179. [crossref]
- Fels DR, Ye J, Segan AT, et al. (2008) Preferential cytotoxicity of bortezomib toward hypoxic tumor cells via overactivation of endoplasmic reticulum stress pathways. Cancer Res 68: 9323-9330.
- Orlowski RZ, Kuhn DJ (2008) Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin Cancer Res 14: 1649-1657. [crossref]
- Li C, Zang Y, Sen M, Leeman-Neill RJ, Man DS, et al. (2009) Bortezomib up-regulates activated signal transducer and activator of transcription-3 and synergizes with inhibitors of signal transducer and activator of transcription-3 to promote head and neck squamous cell carcinoma cell death. Mol Cancer Ther 8: 2211-2220. [crossref]
- Wang Q, He Z, Zhang J, et al. (2005) Overexpression of endoplasmic reticulum molecular chaperone GRP94 and GRP78 in human lung cancer tissues and its significance. Cancer detection and prevention 29: 544-55.
- Song X, Liu X, Chi W, Liu Y, Wei L, et al. (2006) Hypoxia-induced resistance to cisplatin and doxorubicin in non-small cell lung cancer is inhibited by silencing of HIF-1alpha gene. Cancer Chemother Pharmacol 58: 776-784. [crossref]
- Zhou Y, Li M, Wei Y, Feng D, Peng C, et al. (2009) Down-regulation of GRIM-19 expression is associated with hyperactivation of STAT3-induced gene expression and tumor growth in human cervical cancers. J Interferon Cytokine Res 29: 695-703. [crossref]
- Corvinus FM, Orth C, Moriggl R, Tsareva SA, Wagner S, et al. (2005) Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia 7: 545-555. [crossref]
- Yu CL, Jove R and Burakoff SJ (1997): Constitutive activation of the Janus kinase-STAT pathway in T lymphoma overexpressing the Lck protein tyrosine kinase. J Immunol 159: 5206-5210.
- Rahaman SO, Harbor PC, Chernova O, Barnett GH, Vogelbaum MA, et al. (2002) Inhibition of constitutively active Stat3 s.uppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene 21: 8404-8413
- Masuda M, Suzui M, Yasumatu R, et al.(2002) Constitutive activation of signal transducers and activators of transcription 3 correlates with cyclin D1 overexpression and may provide a novel prognostic marker in head and neck squamous cell carcinoma. Cancer Res 62: 3351-3355.
- Horinaga M, Okita H, Nakashima J, Kanao K, Sakamoto M, et al. (2005) Clinical and pathologic significance of activation of signal transducer and activator of transcription 3 in prostate cancer. Urology 66: 671-675.
- Kusaba T, Nakayama T, Yamazumi K, et al. (2005) Expression of p-STAT3 in human colorectal adenocarcinoma and adenoma; correlation with clinicopathological factors. J Clin Pathol 58: 833-838
- Groner B, Lucks P, Borghouts C (2008) The function of Stat3 in tumor cells and their microenvironment. Semin Cell Dev Biol 19: 341-350. [crossref]
- Christoffersen T, Guren TK, Spindler KL, Dahl O, Lonning PE, et al. (2009) Cancer therapy targeted at cellular signal transduction mechanisms: strategies, clinical results, and unresolved issues. Eur J Pharmacol 625: 6-22.
- Hsu HW, Gridley DS, Kim PD, Hu S, de Necochea-Campion R, et al. (2013) Linifanib (ABT-869) enhances radiosensitivity of head and neck squamous cell carcinoma cells. Oral Oncol 49: 591-597. [crossref]
- Sen M, Joyce S, Panahandeh M, Li C, Thomas SM, et al. (2012) Targeting Stat3 abrogates EGFR inhibitor resistance in cancer. Clin Cancer Res 18: 4986-4996. [crossref]
- Rosen DG, Mercado-Uribe I, Yang G, Bast RC Jr, Amin HM, et al. (2006) The role of constitutively active signal transducer and activator of transcription 3 in ovarian tumorigenesis and prognosis. Cancer 107: 2730-2740. [crossref]
- Wang G, Yang ZQ and Zhang K (2010) Endoplasmic reticulum stress response in cancer: molecular mechanism and therapeutic potential. Am J Transl Res 2: 65-74.
- Healy SJ, Gorman AM, Mousavi-Shafaei P, Gupta S and Samali A (2009) Targeting the endoplasmic reticulum-stress response as an anticancer strategy. Eur J Pharmacol 625: 234-246.
- Ishii Y, Pirkmaier A, Alvarez JV, Frank DA, Keselman I, et al. (2006) Cyclin D1 overexpression and response to bortezomib treatment in a breast cancer model. J Natl Cancer Inst 98: 1238-1247. [crossref]
- Zhuang L, Lee CS, Scolyer RA, et al. (2007) Mcl-, Bcl-XL and Stat3 expression are associated with progression of melanoma whereas Bcl-2, AP-2 and MITF levels decrease during progression of melanoma. Mod Pathol 20: 416-426.
- Wang Y, Rishi AK, Puliyappadamba VT, et al. (2010) Targeted proteasome inhibition by Velcade induces apoptosis in human mesothelioma and breast cancer cell lines. Cancer Chemother Pharmacol 66: 455-466.
- Bazzaro M, Lee MK, Zoso A, et al. (2006) Ubiquitin-proteasome system stress sensitizes ovarian cancer to proteasome inhibitor-induced apoptosis. Cancer Res 66: 3754-3763
- Liu T, Peng H, Zhang M, Deng Y and Wu Z (2010) Cucurbitacin B, a small molecule inhibitor of the Stat3 signaling pathway, enhances the chemosensitivity of laryngeal squamous cell carcinoma cells to cisplatin. Eur J Pharmacol 641: 15-22.
- Jia L, Song Q, Zhou C, et al. (2016) Dihydroartemisinin as a Putative STAT3 Inhibitor, Suppresses the Growth of Head and Neck Squamous Cell Carcinoma by Targeting Jak2/STAT3 Signaling. PLoS One 11: e0147157. [crossref]